

Genetics talks to epigenetics? The interplay between sequence variants and chromatin structure
- PMID: 21286314
- PMCID: PMC2945002
- DOI: 10.2174/138920210791616662
Genetics talks to epigenetics? The interplay between sequence variants and chromatin structure
Abstract
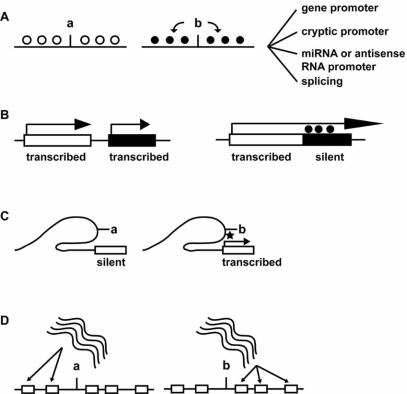
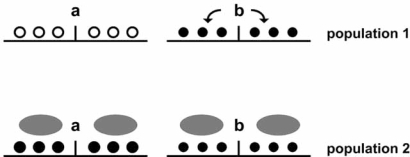
Transcription is regulated by two major mechanisms. On the one hand, changes in DNA sequence are responsible for genetic gene regulation. On the other hand, chromatin structure regulates gene activity at the epigenetic level. Given the fundamental participation of these mechanisms in transcriptional regulation of virtually any gene, they are likely to co-regulate a significant proportion of the genome. The simple concept behind this idea is that a mutation may have a significant impact on local chromatin structure by modifying DNA methylation patterns or histone type recruitment. Yet, the relevance of these interactions is poorly understood. Elucidating how genetic and epigenetic mechanisms co-participate in regulating transcription may assist in some of the unresolved cases of genetic variant-phenotype association. One example is loci that have biologically predictable functions but genotypes that fail to correlate with phenotype, particularly disease outcome. Conversely, a crosstalk between genetics and epigenetics may provide a mechanistic explanation for cases in which a convincing association between phenotype and a genetic variant has been established, but the latter does not lie in a promoter or protein coding sequence. Here, we review recently published data in the field and discuss their implications for genetic variant-phenotype association studies.
Keywords: Chromatin; DNA methylation; epigenetics; genetic variant; histone; single nucleotide polymorphism..
Figures
Similar articles
-
Decoding the epigenetic language of plant development.Mol Plant. 2010 Jul;3(4):719-28. doi: 10.1093/mp/ssq026. Mol Plant. 2010. PMID: 20663898 Free PMC article. Review.
-
Epigenetics: connecting environment and genotype to phenotype and disease.J Dent Res. 2009 May;88(5):400-8. doi: 10.1177/0022034509335868. J Dent Res. 2009. PMID: 19493882 Free PMC article. Review.
-
Interplay between different epigenetic modifications and mechanisms.Adv Genet. 2010;70:101-41. doi: 10.1016/B978-0-12-380866-0.60005-8. Adv Genet. 2010. PMID: 20920747 Review.
-
Retrospective and perspective of plant epigenetics in China.J Genet Genomics. 2018 Nov 20;45(11):621-638. doi: 10.1016/j.jgg.2018.09.004. Epub 2018 Nov 6. J Genet Genomics. 2018. PMID: 30455036 Review.
-
Epigenetics and cell cycle regulation in cystogenesis.Cell Signal. 2020 Apr;68:109509. doi: 10.1016/j.cellsig.2019.109509. Epub 2019 Dec 23. Cell Signal. 2020. PMID: 31874209 Free PMC article. Review.
Cited by
-
The Role of Epigenetic Change in Autism Spectrum Disorders.Front Neurol. 2015 May 26;6:107. doi: 10.3389/fneur.2015.00107. eCollection 2015. Front Neurol. 2015. PMID: 26074864 Free PMC article. Review.
-
Identification of intergenerational epigenetic inheritance by whole genome DNA methylation analysis in trios.Sci Rep. 2023 Dec 2;13(1):21266. doi: 10.1038/s41598-023-48517-3. Sci Rep. 2023. PMID: 38042866 Free PMC article.
-
Genome-Wide DNA Methylation Profile Indicates Potential Epigenetic Regulation of Aging in the Rhesus Macaque Thymus.Int J Mol Sci. 2022 Nov 29;23(23):14984. doi: 10.3390/ijms232314984. Int J Mol Sci. 2022. PMID: 36499310 Free PMC article.
-
Dopamine pathway gene variants may modulate cognitive performance in the DHS - Mind Study.Brain Behav. 2016 Mar 15;6(4):e00446. doi: 10.1002/brb3.446. eCollection 2016 Apr. Brain Behav. 2016. PMID: 27066308 Free PMC article.
-
Maternal genetic variation accounts in part for the associations of maternal size during pregnancy with offspring cardiometabolic risk in adulthood.PLoS One. 2014 Mar 26;9(3):e91835. doi: 10.1371/journal.pone.0091835. eCollection 2014. PLoS One. 2014. PMID: 24670385 Free PMC article.
References
-
- Zhao Z, Fu Y-X, Hewett-Emmett D, Boerwinkle E. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene. 2003;312:207–213. - PubMed
-
- Glinskii AB, Ma J, Ma S, Grant D, Lim CU, Sell S, Glinsky GV. Identification of intergenic trans-regulatory RNAs containing a disease-linked SNP sequence and targeting cell cycle progression/differentiation pathways in multiple common human disorders. Cell Cycle. 2009;8:3925–3942. - PubMed
-
- Yi F, Brubaker PL, Jin T. TCF-4 mediates cell type-specific regulation of proglucagon gene expression by beta-catenin and glycogen synthase kinase-3beta. J. Biol. Chem. 2005;280:1457–1464. - PubMed
-
- Damcott CM, Pollin TI, Reinhart LJ, Ott SH, Shen H, Silver KD, Mitchell BD, Shuldiner AR. Polymorphisms in the transcription factor 7-like 2 (TCF7L2) gene are associated with type 2 diabetes in the Amish: replication and evidence for a role in both insulin secretion and insulin resistance. Diabetes. 2006;55:2654–2659. - PubMed
-
- Saxena R, Gianniny L, Burtt NP, Lyssenko V, Giuducci C, Sjögren M, Florez JC, Almgren P, Isomaa B, Orho-Melander M, Lindblad U, Daly MJ, Tuomi T, Hirschhorn JN, Ardlie KG, Groop LC, Altshuler D. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes. 2006;55:2890–2895. - PubMed
LinkOut - more resources
Full Text Sources