

von Willebrand disease
- PMID: 21289515
- PMCID: PMC3832952
- DOI: 10.1097/GIM.0b013e3182035931
von Willebrand disease
Abstract
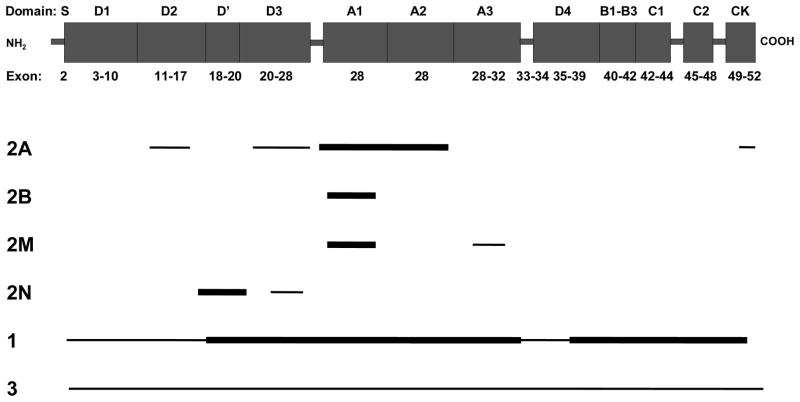
von Willebrand disease is a common inherited bleeding disorder characterized by excessive mucocutaneous bleeding. Characteristic bleeding symptoms include epistaxis, easy bruising, oral cavity bleeding, menorrhagia, bleeding after dental extraction, surgery, and/or childbirth, and in severe cases, bleeding into joints and soft tissues. There are three subtypes: types 1 and 3 represent quantitative variants and type 2 is a group of four qualitative variants: (1) type 2A-characterized by defective von Willebrand factor-dependent platelet adhesion because of decreased high-molecular-weight von Willebrand factor multimers, (2) type 2B-caused by pathologically increased von Willebrand factor-platelet interactions, (3) type 2M-caused by decreased von Willebrand factor-platelet interactions not based on the loss of high-molecular-weight multimers, and (4) type 2N-characterized by reduced binding of von Willebrand factor to factor VIII. The diagnosis of von Willebrand disease requires specialized assays of von Willebrand factor and/or molecular genetic testing of von Willebrand factor. Severe bleeding episodes can be prevented or controlled with intravenous infusions of virally inactivated plasma-derived clotting factor concentrates containing both von Willebrand factor and factor VIII. Depending on the von Willebrand disease type, mild bleeding episodes usually respond to intravenous or subcutaneous treatment with desmopressin, a vasopressin analog. Other treatments that can reduce symptoms include fibrinolytic inhibitors and hormones for menorrhagia.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest
Figures
References
-
- von Willebrand EA. Hereditar pseudohemofili. Finska Lakarsallskapets Handl. 1926;67:7–112.
-
- Lynch DC, Zimmerman TS, Collins CJ, et al. Molecular cloning of cDNA for human von Willebrand factor: authentication by a new method. Cell. 1985;41:49–56. - PubMed
-
- Ginsburg D, Handin RI, Bonthron DT, et al. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science. 1985;228:1401–6. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
