

Structural variation in the chicken genome identified by paired-end next-generation DNA sequencing of reduced representation libraries
- PMID: 21291514
- PMCID: PMC3039614
- DOI: 10.1186/1471-2164-12-94
Structural variation in the chicken genome identified by paired-end next-generation DNA sequencing of reduced representation libraries
Abstract
Background: Variation within individual genomes ranges from single nucleotide polymorphisms (SNPs) to kilobase, and even megabase, sized structural variants (SVs), such as deletions, insertions, inversions, and more complex rearrangements. Although much is known about the extent of SVs in humans and mice, species in which they exert significant effects on phenotypes, very little is known about the extent of SVs in the 2.5-times smaller and less repetitive genome of the chicken.
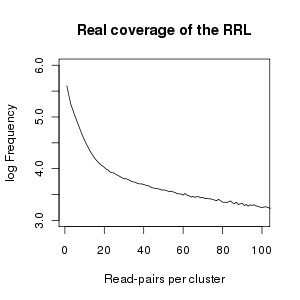
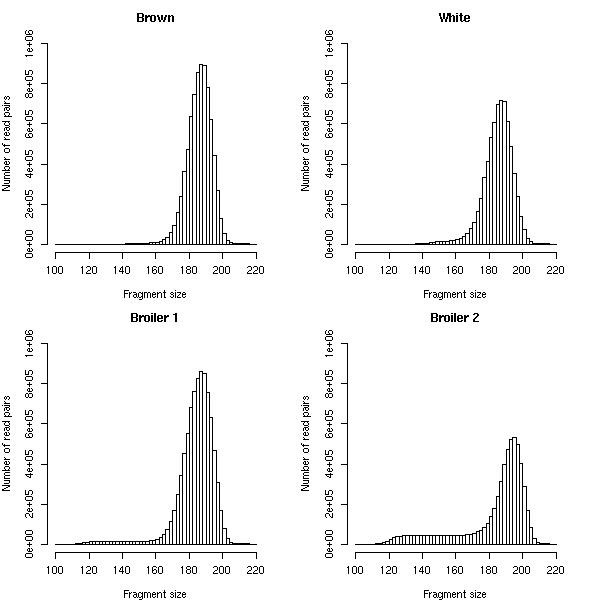
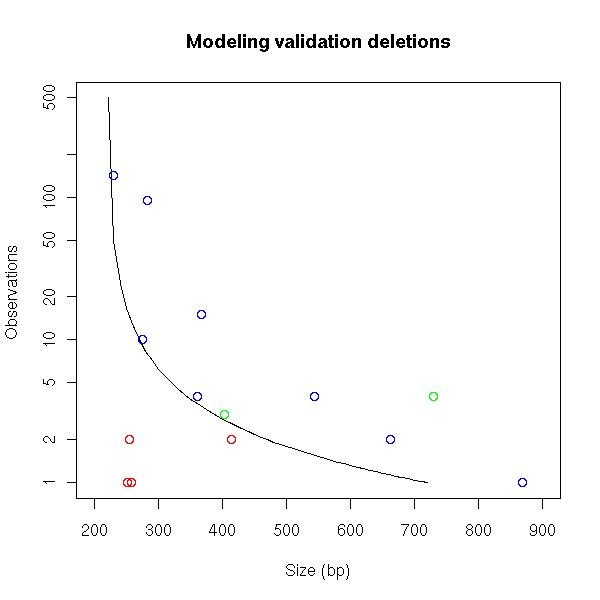
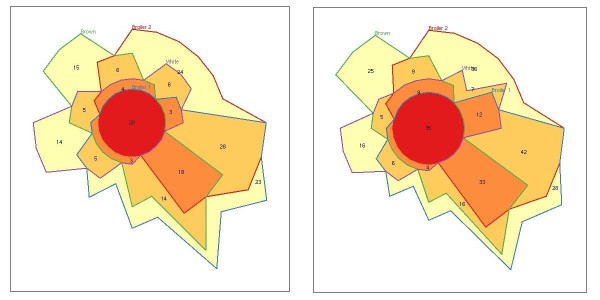
Results: We identified hundreds of shared and divergent SVs in four commercial chicken lines relative to the reference chicken genome. The majority of SVs were found in intronic and intergenic regions, and we also found SVs in the coding regions. To identify the SVs, we combined high-throughput short read paired-end sequencing of genomic reduced representation libraries (RRLs) of pooled samples from 25 individuals and computational mapping of DNA sequences from a reference genome.
Conclusion: We provide a first glimpse of the high abundance of small structural genomic variations in the chicken. Extrapolating our results, we estimate that there are thousands of rearrangements in the chicken genome, the majority of which are located in non-coding regions. We observed that structural variation contributes to genetic differentiation among current domesticated chicken breeds and the Red Jungle Fowl. We expect that, because of their high abundance, SVs might explain phenotypic differences and play a role in the evolution of the chicken genome. Finally, our study exemplifies an efficient and cost-effective approach for identifying structural variation in sequenced genomes.
Figures

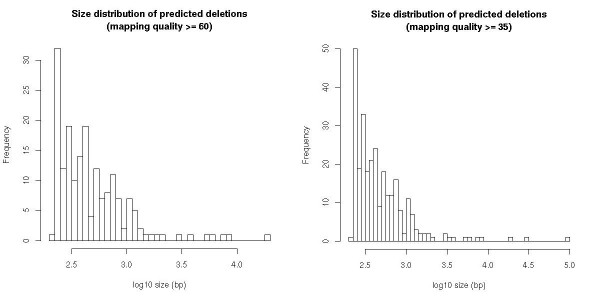
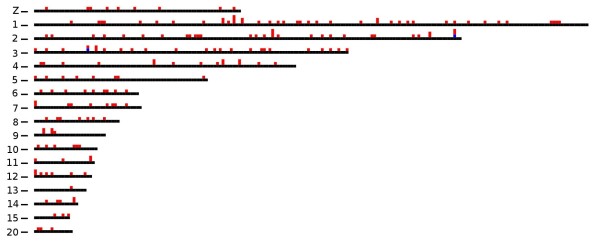

References
-
- McKernan KJ, Peckham HE, Costa G, McLaughlin S, Tsung E, Fu Y, Clouser C, Dunkan C, Ichikawa J, Lee C, Zhang Z, Sheridan A, Fu H, Ranade S, Dimilanta E, Sokolsky T, Zhang L, Hendrickson C, Li B, Kotler L, Stuart J, Malek J, Manning J, Antipova A, Perez D, Moore M, Hayashibara K, Lyons M, Beaudoin R, Coleman B, Laptewicz M, Sanicandro A, Rhodes M, Vega FDL, Gottimukkala RK, Hyland F, Reese M, Yang S, Bafna V, Bashir A, Macbride A, Aklan C, Kidd JM, Eichler EE, Blanchard AP. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. - DOI - PMC - PubMed
-
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders ACE, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. - DOI - PMC - PubMed
-
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tüzün E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M, Smith JD, Korn JM, McCarroll SA, Altshuler DA, Peiffer DA, Dorschner M, Stamatoyannopoulos J, Schwartz D, Nickerson DA, Mullikin JC, Wilson RK, Bruhn L, Olson MV, Kaul R, Smith DR, Eichler EE. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
