

Modulating micro-opioid receptor phosphorylation switches agonist-dependent signaling as reflected in PKCepsilon activation and dendritic spine stability
- PMID: 21292762
- PMCID: PMC3069472
- DOI: 10.1074/jbc.M110.177089
Modulating micro-opioid receptor phosphorylation switches agonist-dependent signaling as reflected in PKCepsilon activation and dendritic spine stability
Abstract
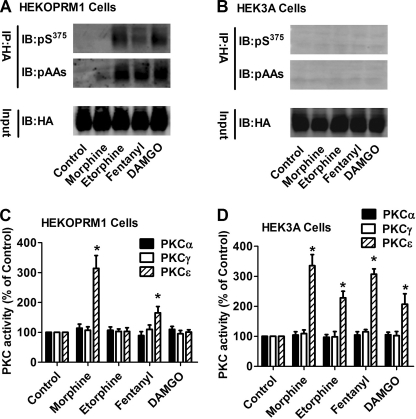
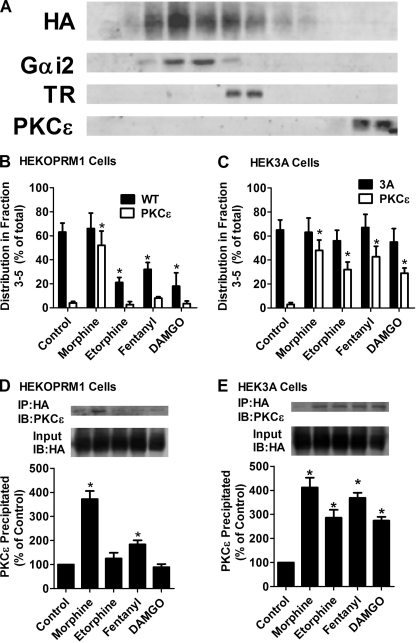
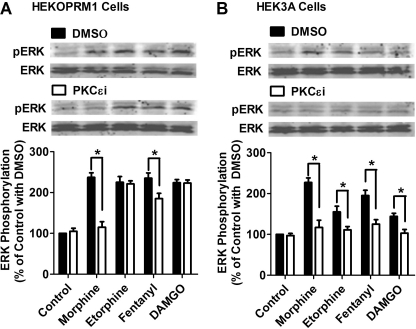
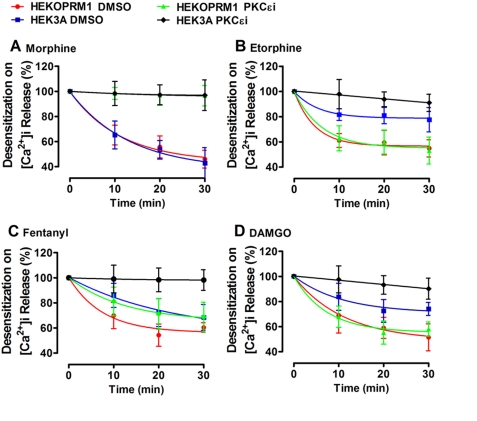
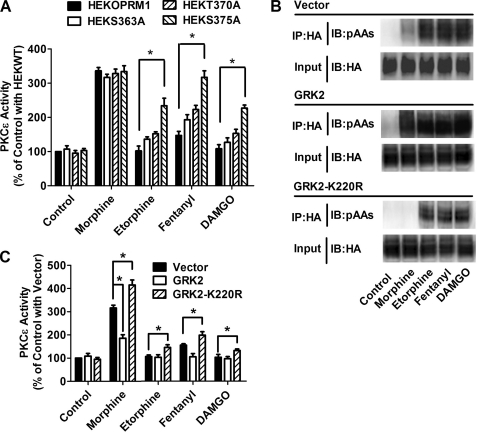
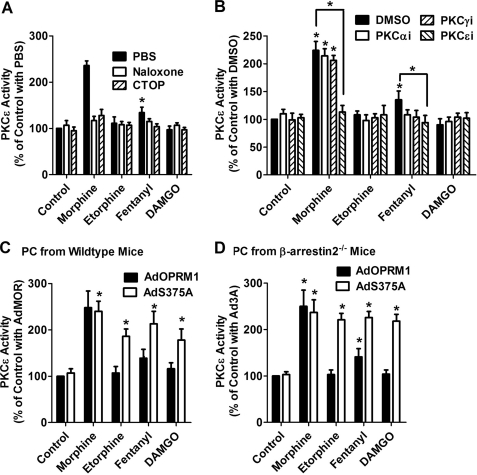
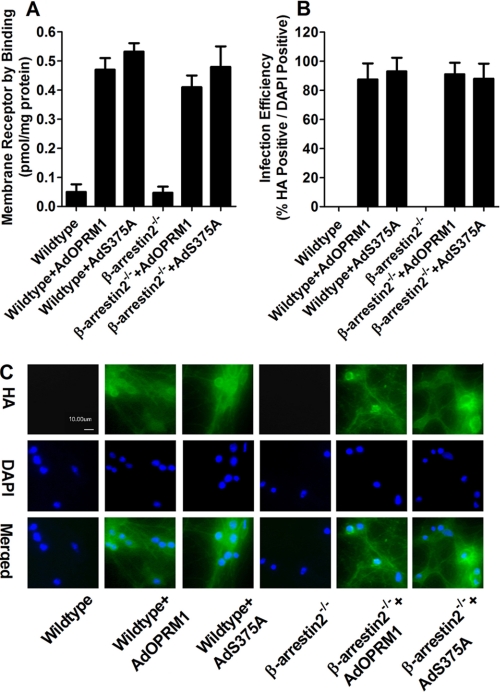
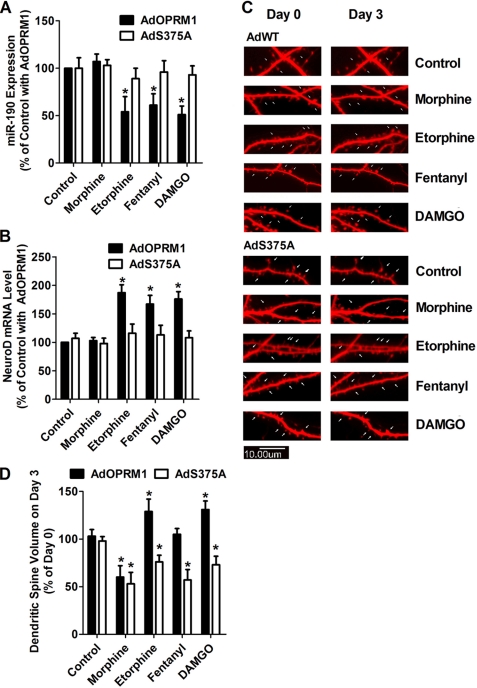
A new role of G protein-coupled receptor (GPCR) phosphorylation was demonstrated in the current studies by using the μ-opioid receptor (OPRM1) as a model. Morphine induces a low level of receptor phosphorylation and uses the PKCε pathway to induce ERK phosphorylation and receptor desensitization, whereas etorphine, fentanyl, and [D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) induce extensive receptor phosphorylation and use the β-arrestin2 pathway. Blocking OPRM1 phosphorylation (by mutating Ser363, Thr370 and Ser375 to Ala) enabled etorphine, fentanyl, and DAMGO to use the PKCε pathway. This was not due to the decreased recruitment of β-arrestin2 to the receptor signaling complex, because these agonists were unable to use the PKCε pathway when β-arrestin2 was absent. In addition, overexpressing G protein-coupled receptor kinase 2 (GRK2) decreased the ability of morphine to activate PKCε, whereas overexpressing dominant-negative GRK2 enabled etorphine, fentanyl, and DAMGO to activate PKCε. Furthermore, by overexpressing wild-type OPRM1 and a phosphorylation-deficient mutant in primary cultures of hippocampal neurons, we demonstrated that receptor phosphorylation contributes to the differential effects of agonists on dendritic spine stability. Phosphorylation blockage made etorphine, fentanyl, and DAMGO function as morphine in the primary cultures. Therefore, agonist-dependent phosphorylation of GPCR regulates the activation of the PKC pathway and the subsequent responses.
Figures
References
-
- Urban J. D., Clarke W. P., von Zastrow M., Nichols D. E., Kobilka B., Weinstein H., Javitch J. A., Roth B. L., Christopoulos A., Sexton P. M., Miller K. J., Spedding M., Mailman R. B. (2007) J. Pharmacol. Exp. Ther. 320, 1–13 - PubMed
-
- Violin J. D., Lefkowitz R. J. (2007) Trends Pharmacol. Sci. 28, 416–422 - PubMed
-
- DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) Annu. Rev. Physiol. 69, 483–510 - PubMed
-
- Kobayashi H., Narita Y., Nishida M., Kurose H. (2005) Cell Signal. 17, 1248–1253 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
