

Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines
- PMID: 21295703
- PMCID: PMC3063454
- DOI: 10.1016/j.cell.2010.12.032
Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines
Abstract
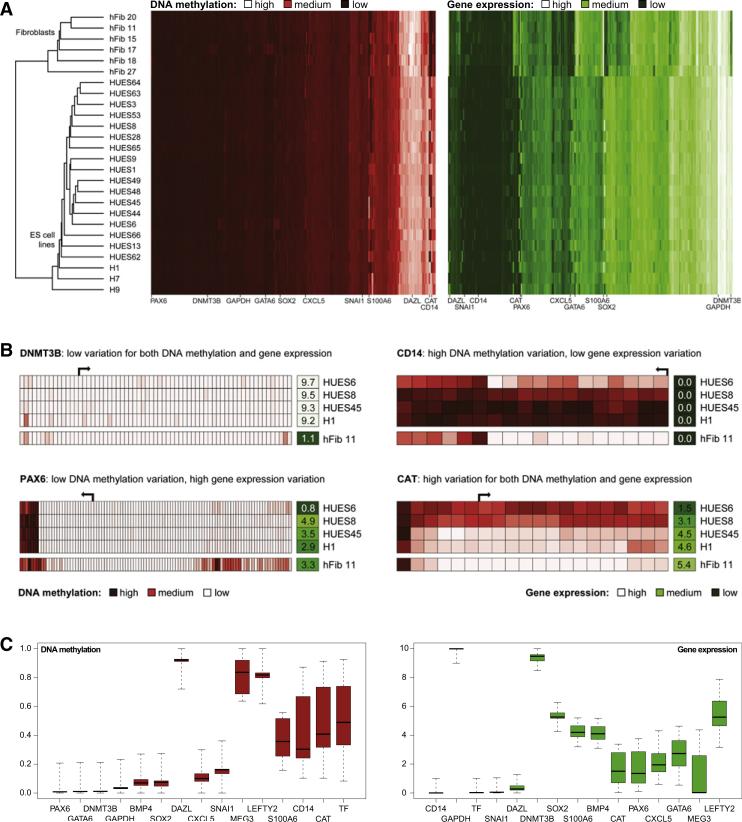
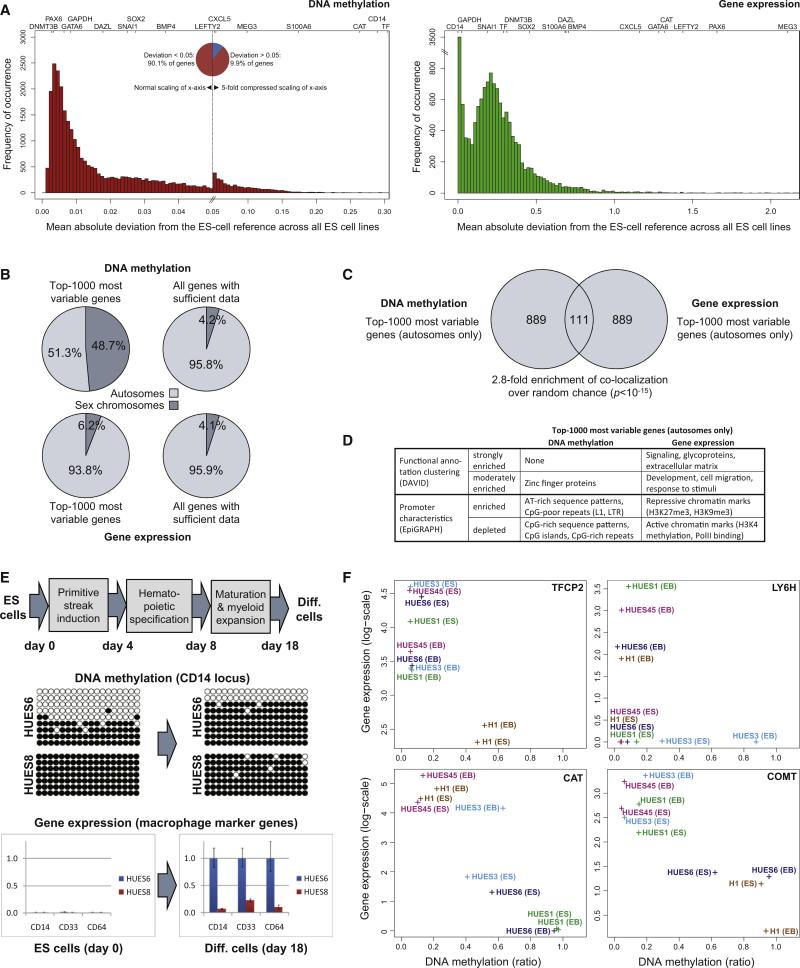
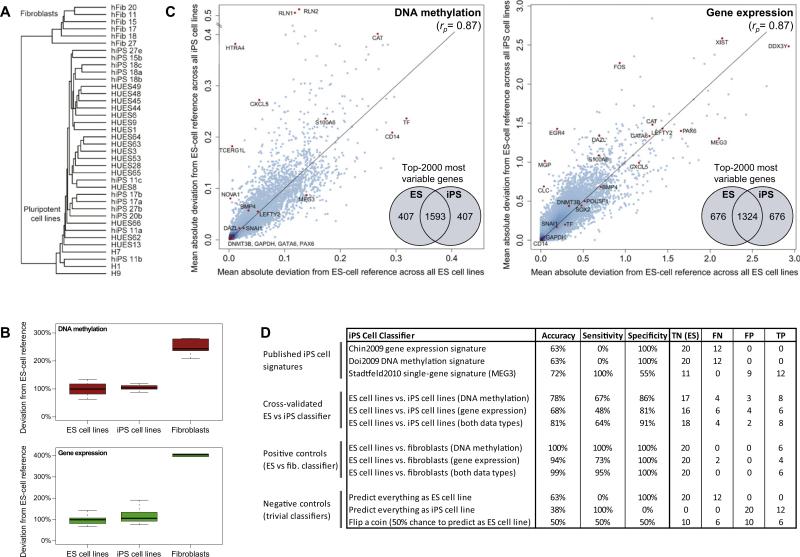
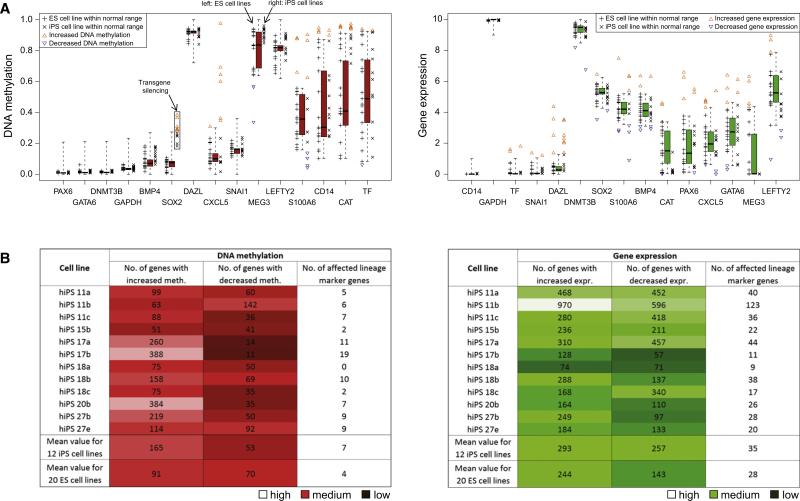
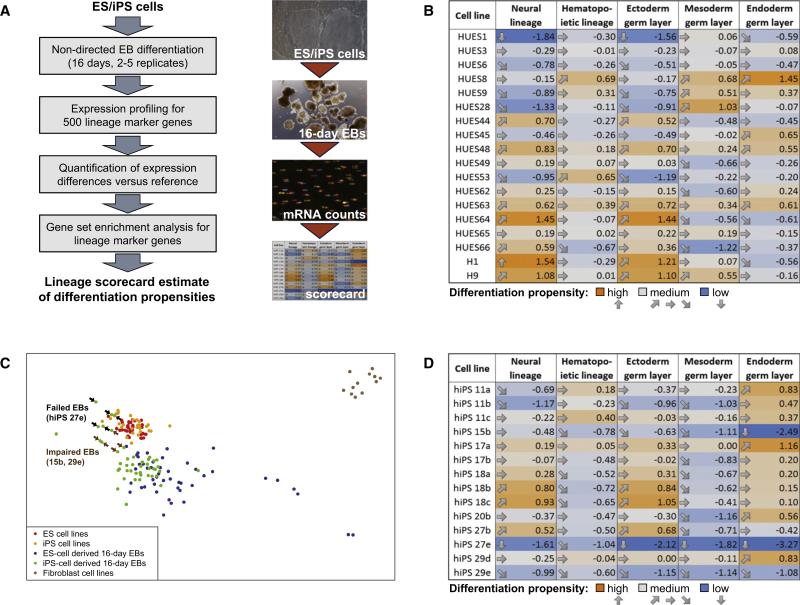
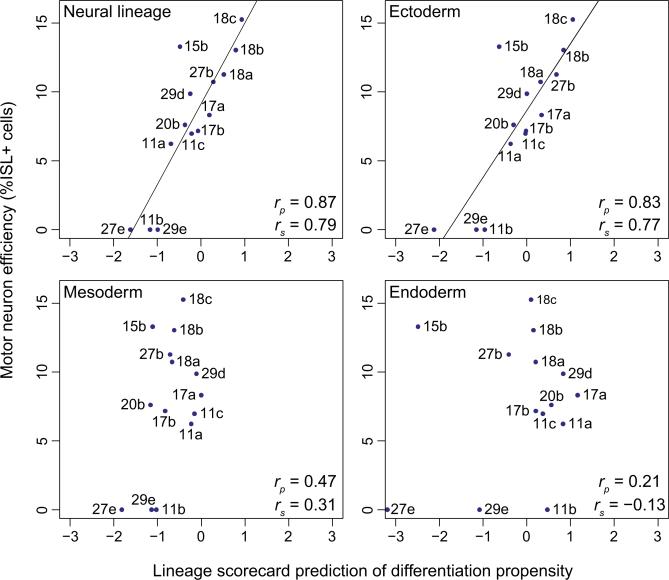
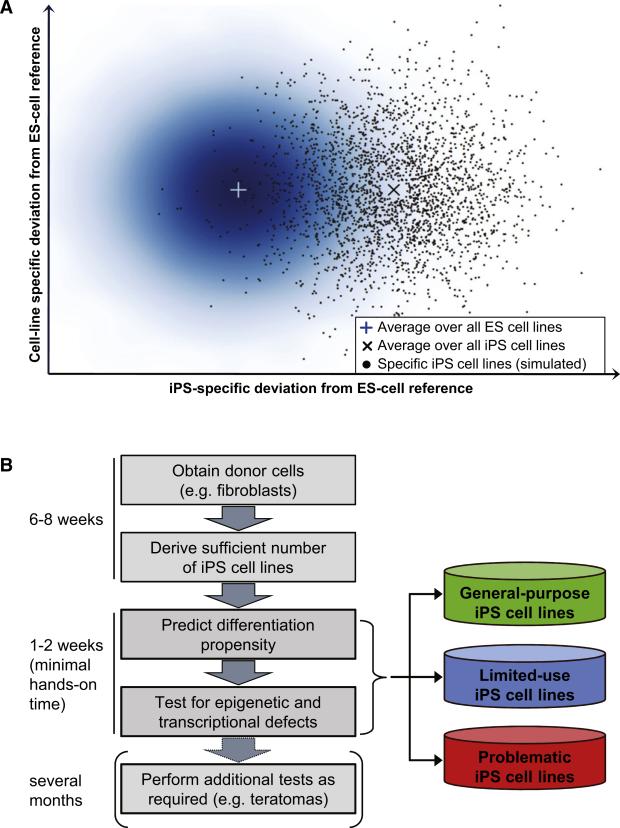
The developmental potential of human pluripotent stem cells suggests that they can produce disease-relevant cell types for biomedical research. However, substantial variation has been reported among pluripotent cell lines, which could affect their utility and clinical safety. Such cell-line-specific differences must be better understood before one can confidently use embryonic stem (ES) or induced pluripotent stem (iPS) cells in translational research. Toward this goal we have established genome-wide reference maps of DNA methylation and gene expression for 20 previously derived human ES lines and 12 human iPS cell lines, and we have measured the in vitro differentiation propensity of these cell lines. This resource enabled us to assess the epigenetic and transcriptional similarity of ES and iPS cells and to predict the differentiation efficiency of individual cell lines. The combination of assays yields a scorecard for quick and comprehensive characterization of pluripotent cell lines.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Stem cells: Scorecards for pluripotent cell lines.Nat Rev Genet. 2011 Mar;12(3):153. doi: 10.1038/nrg2961. Nat Rev Genet. 2011. PMID: 21331086 No abstract available.
-
Testing pluripotency.Nat Methods. 2011 Apr;8(4):287. doi: 10.1038/nmeth0411-287. Nat Methods. 2011. PMID: 21574272 No abstract available.
References
-
- Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, Bello PA, Benvenisty N, Berry LS, Bevan S, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007;25:803–816. - PubMed
-
- Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat. Rev. Genet. 2006;7:55–65. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
