

Modeling protein structure at near atomic resolutions with Gorgon
- PMID: 21296162
- PMCID: PMC3078171
- DOI: 10.1016/j.jsb.2011.01.015
Modeling protein structure at near atomic resolutions with Gorgon
Abstract
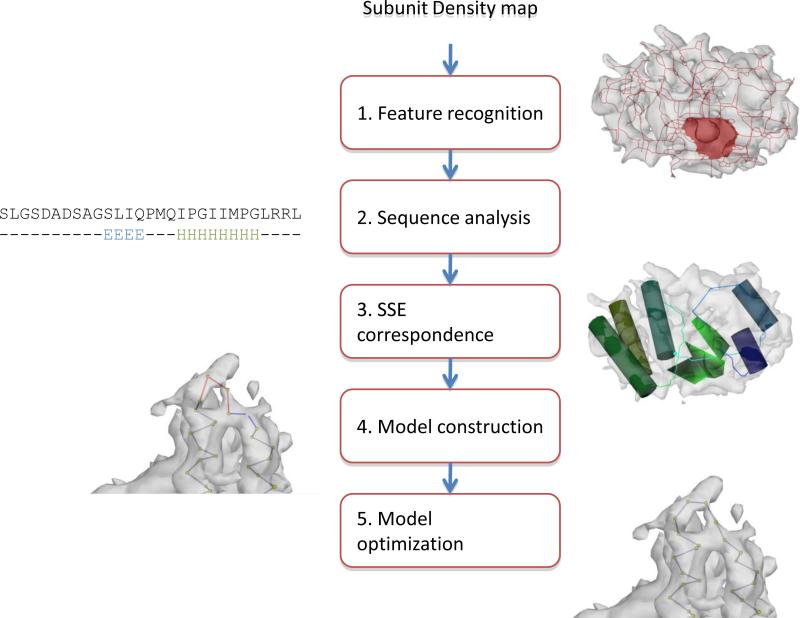
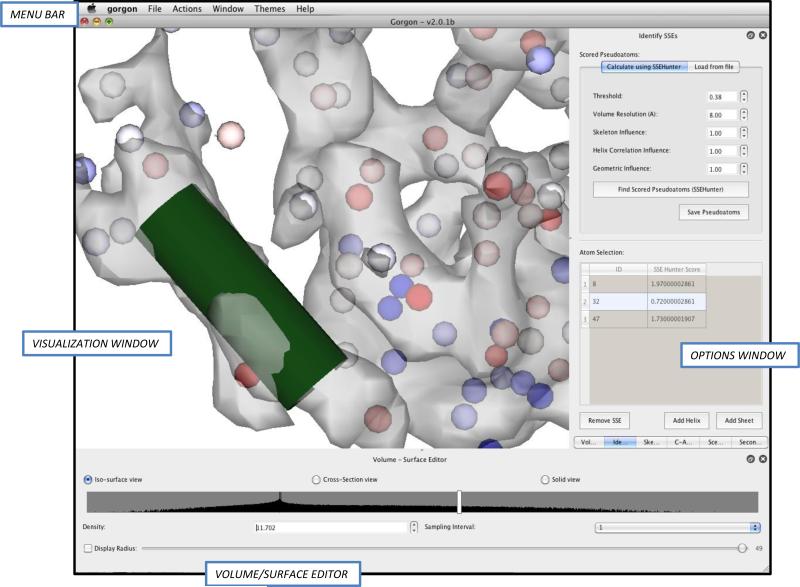
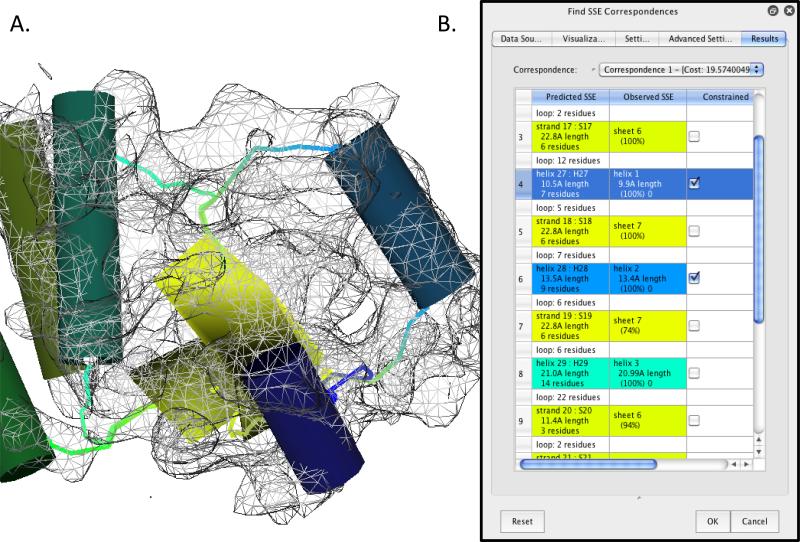
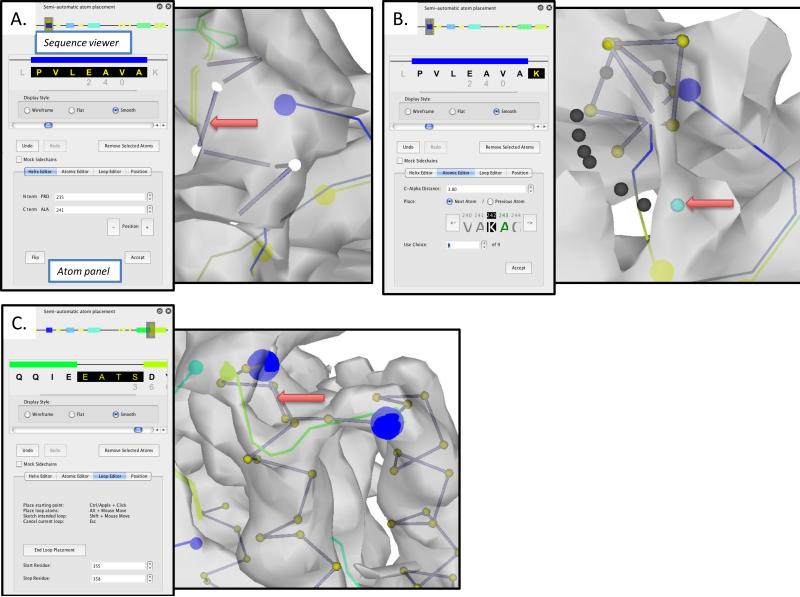
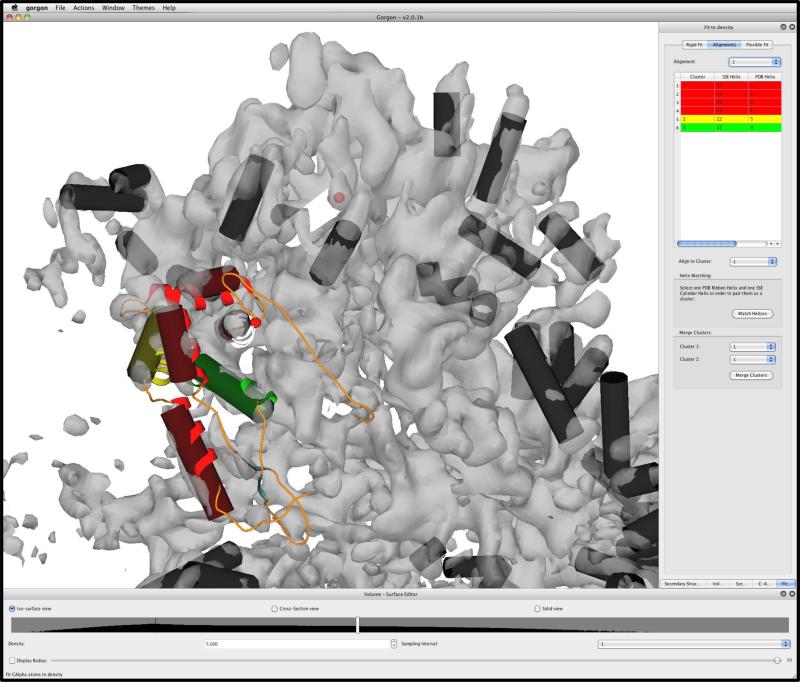
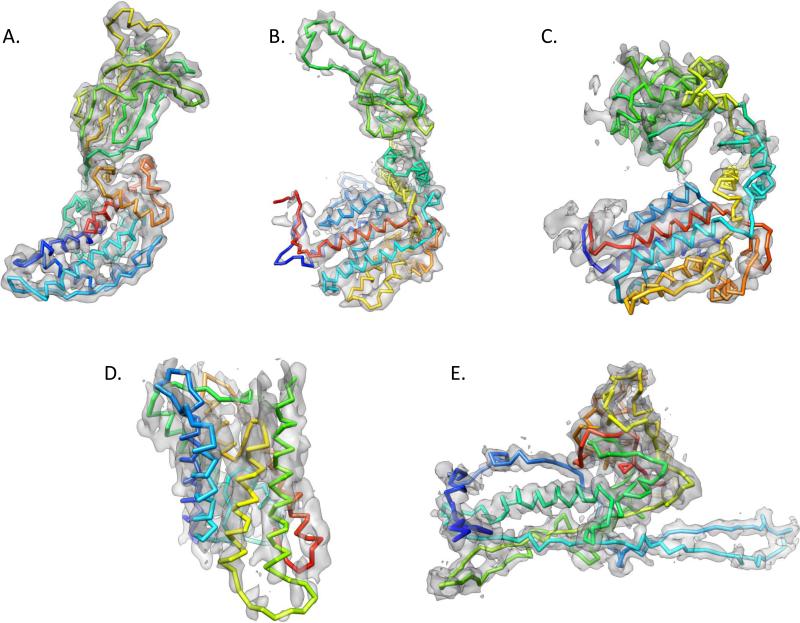
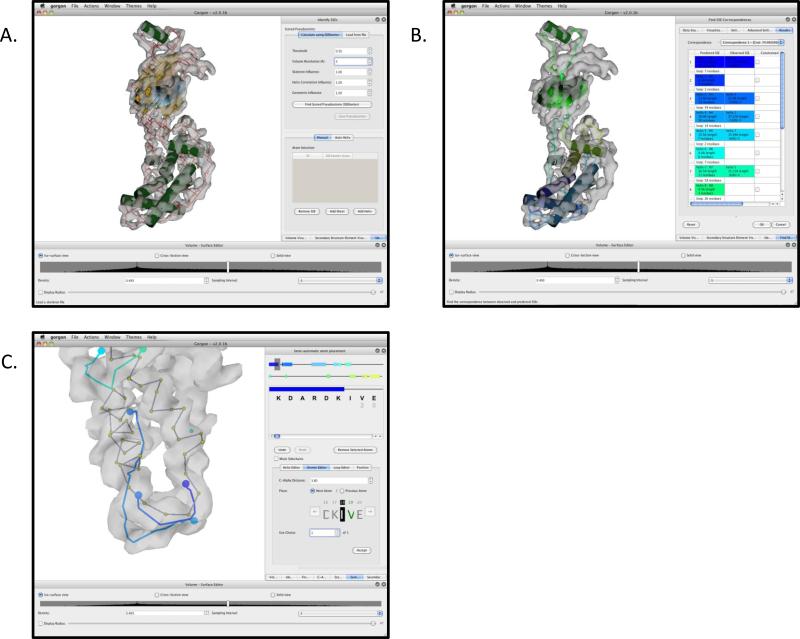
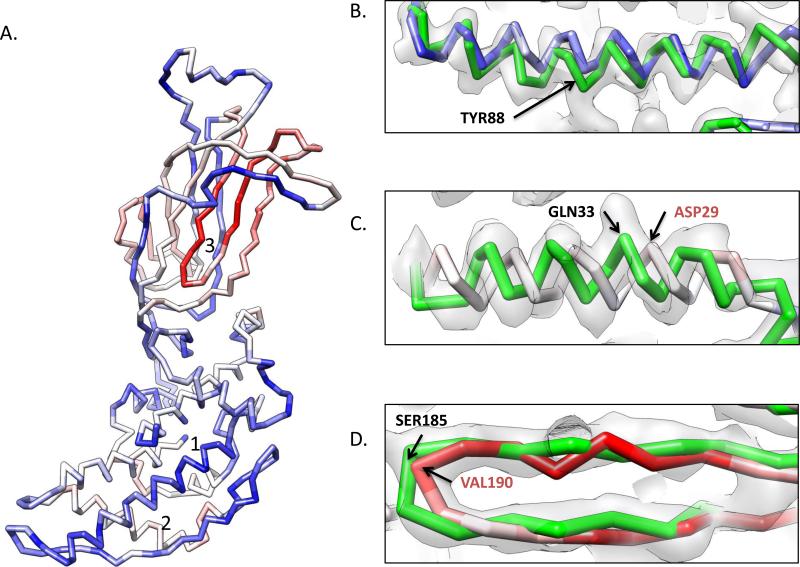
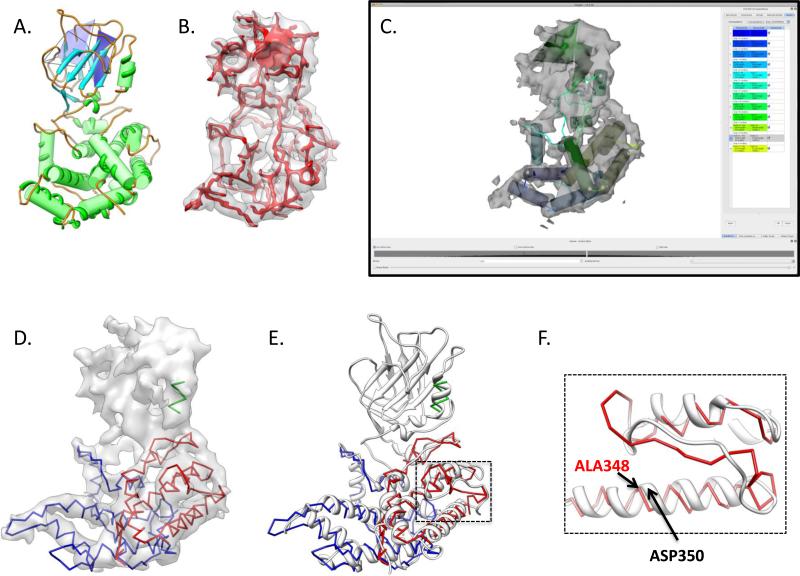
Electron cryo-microscopy (cryo-EM) has played an increasingly important role in elucidating the structure and function of macromolecular assemblies in near native solution conditions. Typically, however, only non-atomic resolution reconstructions have been obtained for these large complexes, necessitating computational tools for integrating and extracting structural details. With recent advances in cryo-EM, maps at near-atomic resolutions have been achieved for several macromolecular assemblies from which models have been manually constructed. In this work, we describe a new interactive modeling toolkit called Gorgon targeted at intermediate to near-atomic resolution density maps (10-3.5 Å), particularly from cryo-EM. Gorgon's de novo modeling procedure couples sequence-based secondary structure prediction with feature detection and geometric modeling techniques to generate initial protein backbone models. Beyond model building, Gorgon is an extensible interactive visualization platform with a variety of computational tools for annotating a wide variety of 3D volumes. Examples from cryo-EM maps of Rotavirus and Rice Dwarf Virus are used to demonstrate its applicability to modeling protein structure.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Abeysinghe S, Ju T, Baker ML, Chiu W. Shape modeling and matching in identifying 3D protein structures. COMPUT AIDED DESIGN. 2008a;40:708–720.
-
- Abeysinghe SS, Baker ML, Chiu W, Ju T. Segmentation-free skeletonization of grayscale volumes for shape understanding. IEEE International Conference on Shape Modeling and Applications. 2008b:63–71.
-
- Abeysinghe SS, Ju T. Interactive skeletonization of intensity volumes. The Visual Compter. 2009;25:627–635.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
