

Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution
- PMID: 21298013
- PMCID: PMC3029335
- DOI: 10.1371/journal.pone.0016329
Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution
Abstract
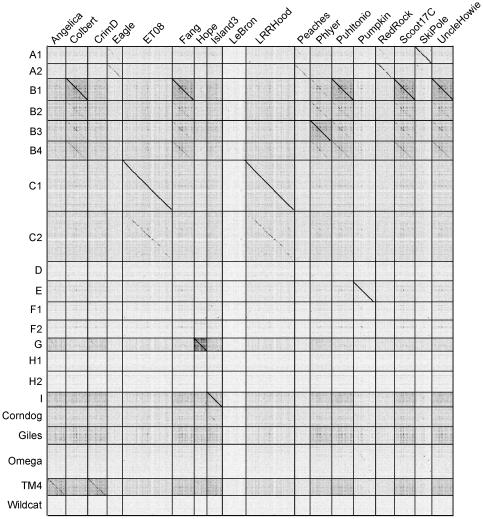
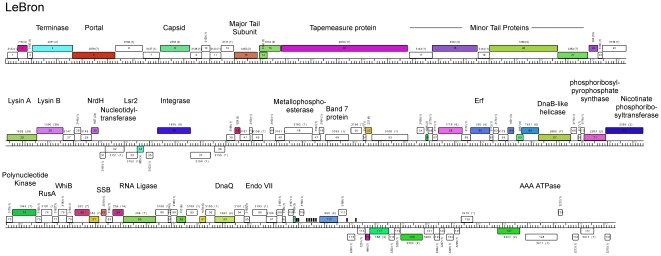
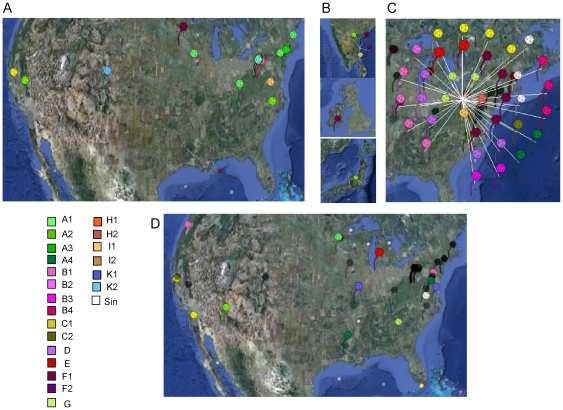
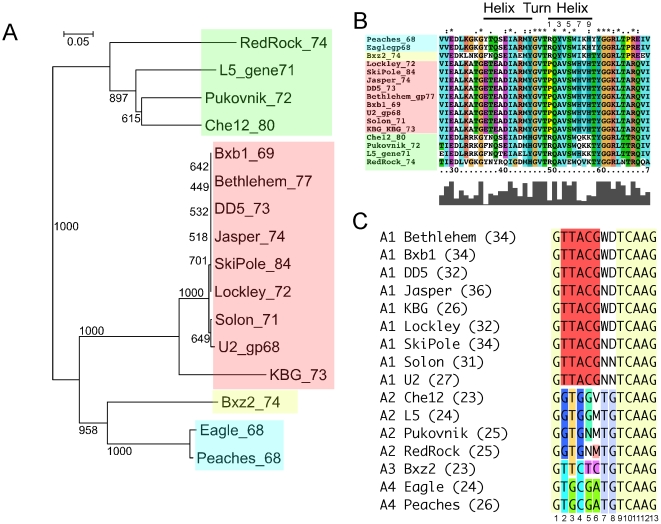
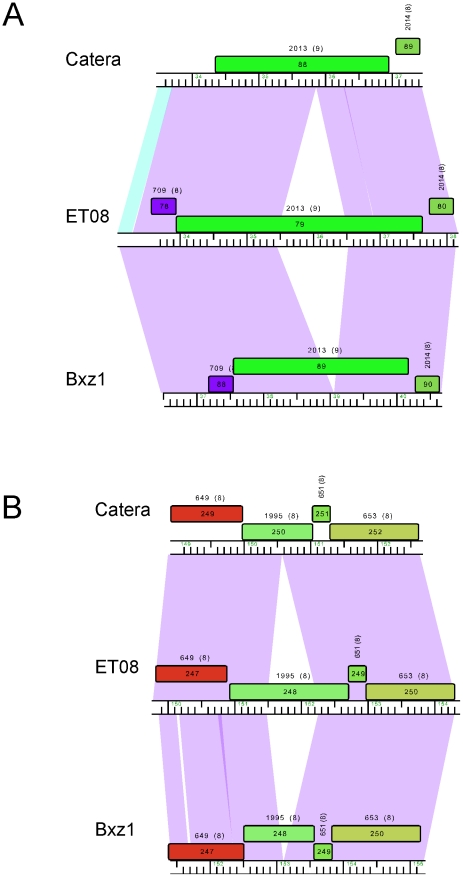
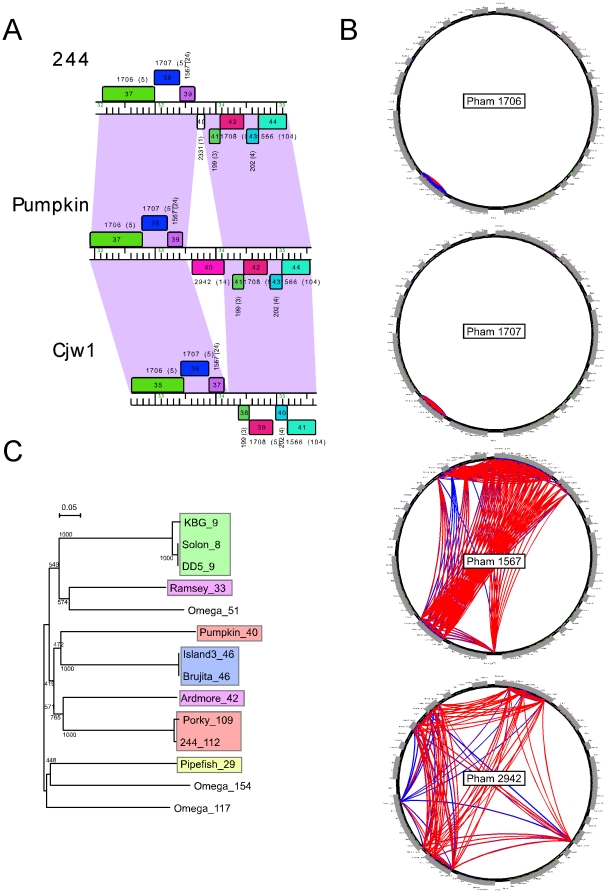
Mycobacteriophages are viruses that infect mycobacterial hosts such as Mycobacterium smegmatis and Mycobacterium tuberculosis. All mycobacteriophages characterized to date are dsDNA tailed phages, and have either siphoviral or myoviral morphotypes. However, their genetic diversity is considerable, and although sixty-two genomes have been sequenced and comparatively analyzed, these likely represent only a small portion of the diversity of the mycobacteriophage population at large. Here we report the isolation, sequencing and comparative genomic analysis of 18 new mycobacteriophages isolated from geographically distinct locations within the United States. Although no clear correlation between location and genome type can be discerned, these genomes expand our knowledge of mycobacteriophage diversity and enhance our understanding of the roles of mobile elements in viral evolution. Expansion of the number of mycobacteriophages grouped within Cluster A provides insights into the basis of immune specificity in these temperate phages, and we also describe a novel example of apparent immunity theft. The isolation and genomic analysis of bacteriophages by freshman college students provides an example of an authentic research experience for novice scientists.
Conflict of interest statement
Figures





References
-
- Suttle CA. Marine viruses—major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–812. - PubMed
-
- Hendrix RW. Bacteriophage genomics. Curr Opin Microbiol. 2003;6:506–511. - PubMed
-
- Brussow H, Hendrix RW. Phage genomics: small is beautiful. Cell. 2002;108:13–16. - PubMed
-
- Hyman P, Abedon ST. Bacteriophage host range and bacterial resistance. Adv Appl Microbiol. 2010;70:217–248. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
