

A novel role for ATM in regulating proteasome-mediated protein degradation through suppression of the ISG15 conjugation pathway
- PMID: 21298066
- PMCID: PMC3027683
- DOI: 10.1371/journal.pone.0016422
A novel role for ATM in regulating proteasome-mediated protein degradation through suppression of the ISG15 conjugation pathway
Abstract
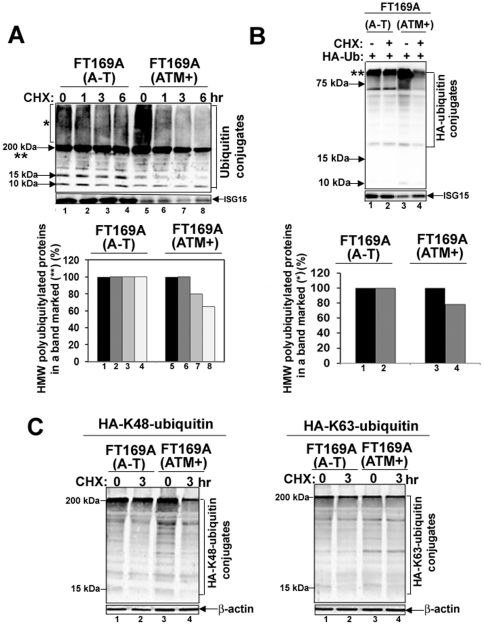
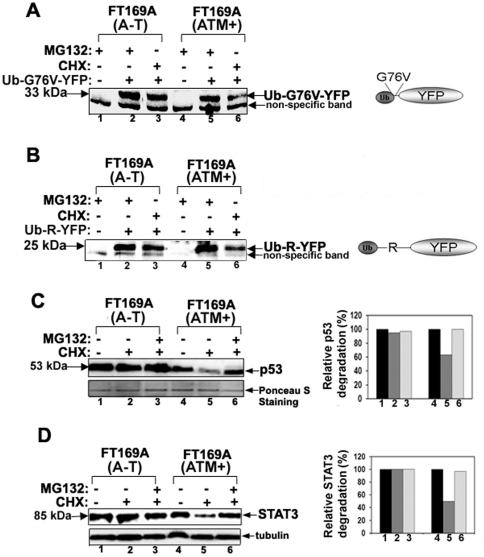
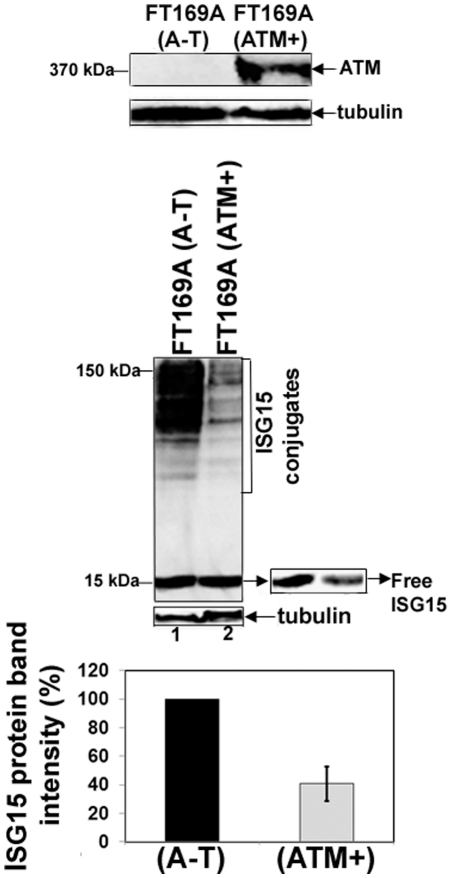
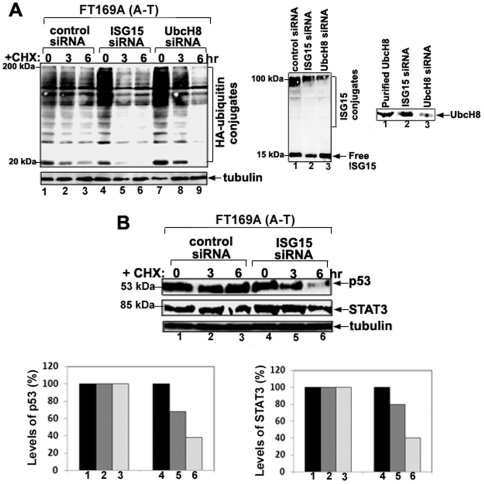
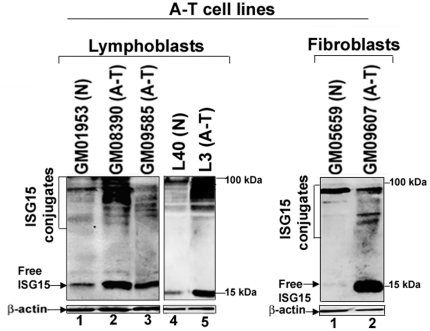
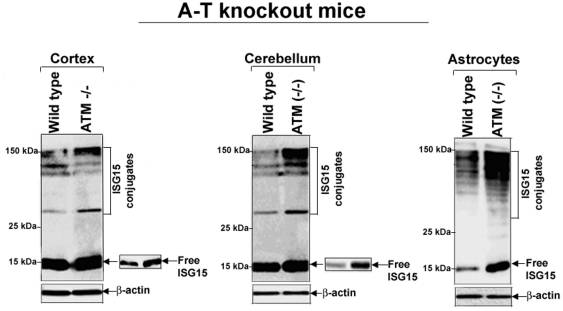
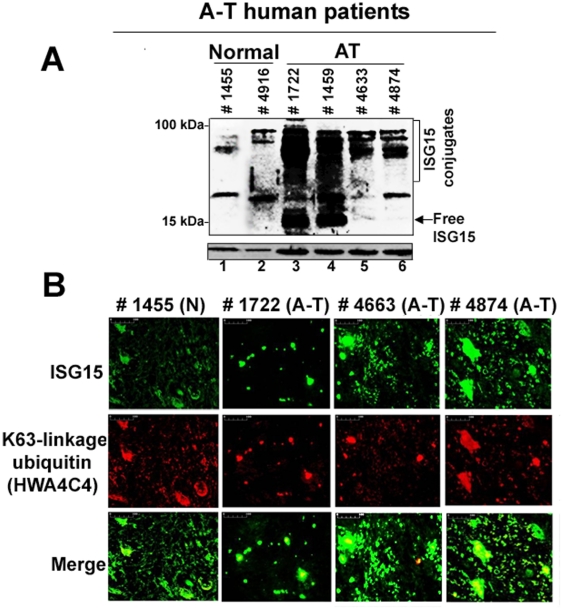
Ataxia Telangiectasia (A-T) is an inherited immunodeficiency disorder wherein mutation of the ATM kinase is responsible for the A-T pathogenesis. Although the precise role of ATM in A-T pathogenesis is still unclear, its function in responding to DNA damage has been well established. Here we demonstrate that in addition to its role in DNA repair, ATM also regulates proteasome-mediated protein turnover through suppression of the ISG15 pathway. This conclusion is based on three major pieces of evidence: First, we demonstrate that proteasome-mediated protein degradation is impaired in A-T cells. Second, we show that the reduced protein turnover is causally linked to the elevated expression of the ubiquitin-like protein ISG15 in A-T cells. Third, we show that expression of the ISG15 is elevated in A-T cells derived from various A-T patients, as well as in brain tissues derived from the ATM knockout mice and A-T patients, suggesting that ATM negatively regulates the ISG15 pathway. Our current findings suggest for the first time that proteasome-mediated protein degradation is impaired in A-T cells due to elevated expression of the ISG15 conjugation pathway, which could contribute to progressive neurodegeneration in A-T patients.
Conflict of interest statement
Figures
References
-
- Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187–1196. - PubMed
-
- Lavin MF, Scott S, Gueven N, Kozlov S, Peng C, et al. Functional consequences of sequence alterations in the ATM gene. DNA Repair (Amst) 2004;3:1197–1205. - PubMed
-
- Boder E. Ataxia-telangiectasia: an overview. Kroc Found Ser. 1985;19:1–63. - PubMed
-
- Jentsch S, Pyrowolakis G. Ubiquitin and its kin: how close are the family ties? Trends Cell Biol. 2000;10:335–342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
