

CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity
- PMID: 21300915
- PMCID: PMC3039853
- DOI: 10.1084/jem.20102010
CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity
Abstract
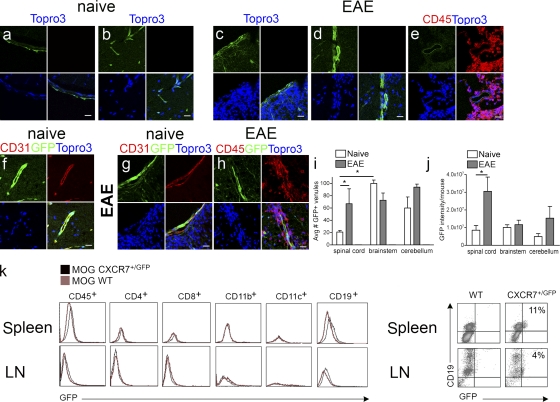
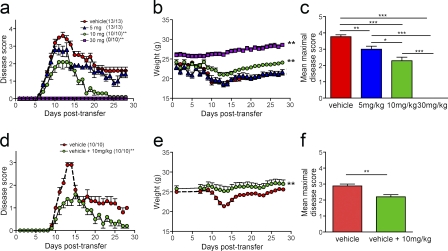
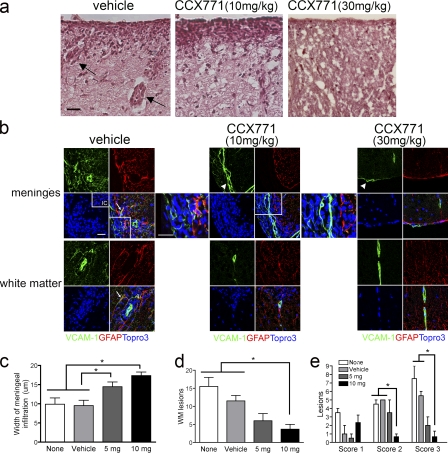
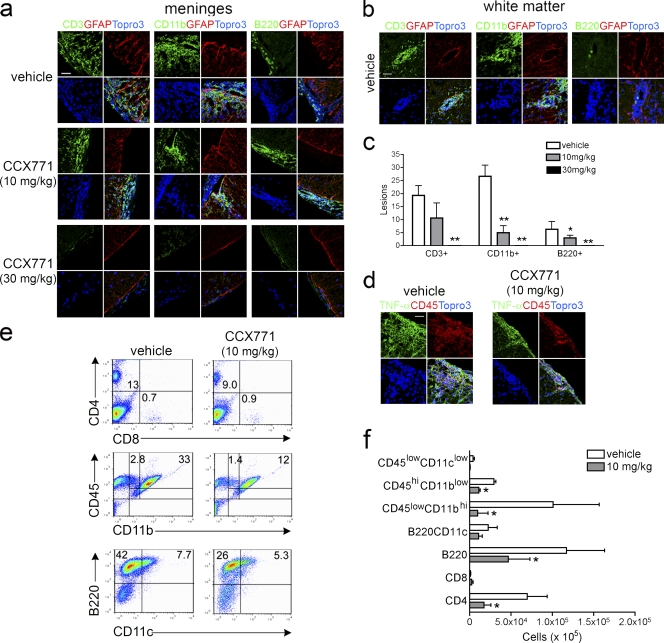
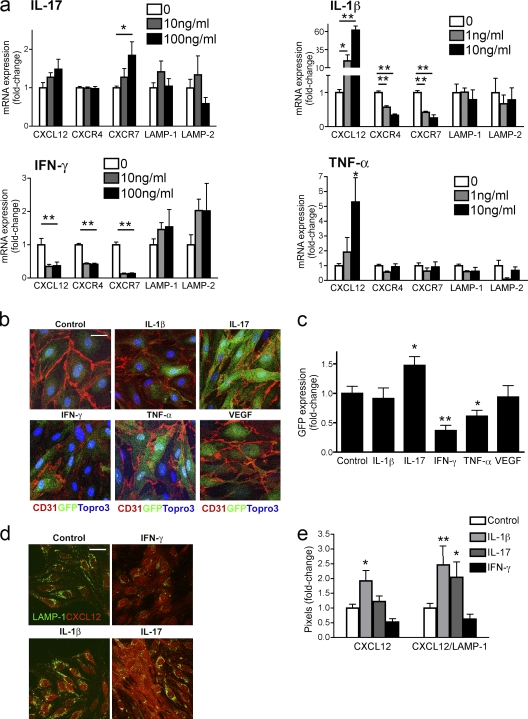
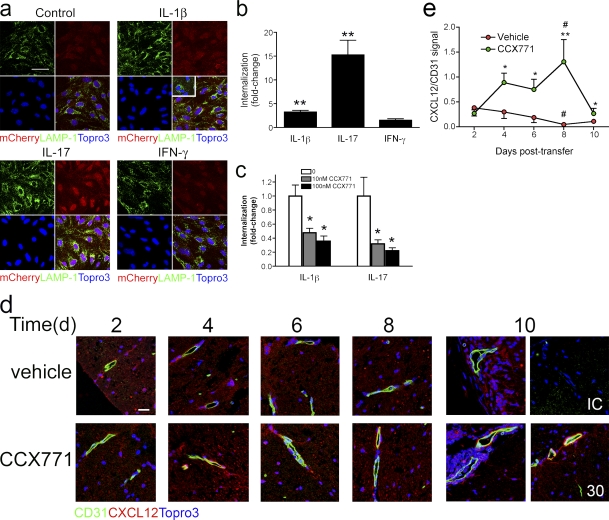
Loss of CXCL12, a leukocyte localizing cue, from abluminal surfaces of the blood-brain barrier occurs in multiple sclerosis (MS) lesions. However, the mechanisms and consequences of reduced abluminal CXCL12 abundance remain unclear. Here, we show that activation of CXCR7, which scavenges CXCL12, is essential for leukocyte entry via endothelial barriers into the central nervous system (CNS) parenchyma during experimental autoimmune encephalomyelitis (EAE), a model for MS. CXCR7 expression on endothelial barriers increased during EAE at sites of inflammatory infiltration. Treatment with a CXCR7 antagonist ameliorated EAE, reduced leukocyte infiltration into the CNS parenchyma and parenchymal VCAM-1 expression, and increased abluminal levels of CXCL12. Interleukin 17 and interleukin 1β increased, whereas interferon-γ decreased, CXCR7 expression on and CXCL12 internalization in primary brain endothelial cells in vitro. These findings identify molecular requirements for the transvascular entry of leukocytes into the CNS and suggest that CXCR7 blockade may have therapeutic utility for the treatment of MS.
Figures
References
-
- Afonso P.V., Ozden S., Prevost M.C., Schmitt C., Seilhean D., Weksler B., Couraud P.O., Gessain A., Romero I.A., Ceccaldi P.E. 2007. Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J. Immunol. 179:2576–2583 - PubMed
-
- Argaw A.T., Zhang Y., Snyder B.J., Zhao M.L., Kopp N., Lee S.C., Raine C.S., Brosnan C.F., John G.R. 2006. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 177:5574–5584 - PubMed
-
- Axtell R.C., de Jong B.A., Boniface K., van der Voort L.F., Bhat R., De Sarno P., Naves R., Han M., Zhong F., Castellanos J.G., et al. 2010. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat. Med. 16:406–412 10.1038/nm.2110 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
