

Review
doi: 10.1038/nrg2950.
Epub 2011 Feb 8.
The importance of phase information for human genomics
Affiliations
- PMID: 21301473
- PMCID: PMC3753045
- DOI: 10.1038/nrg2950
Item in Clipboard
Review
The importance of phase information for human genomics
Nat Rev Genet.
2011 Mar.
Abstract
Contemporary sequencing studies often ignore the diploid nature of the human genome because they do not routinely separate or 'phase' maternally and paternally derived sequence information. However, many findings - both from recent studies and in the more established medical genetics literature - indicate that relationships between human DNA sequence and phenotype, including disease, can be more fully understood with phase information. Thus, the existing technological impediments to obtaining phase information must be overcome if human genomics is to reach its full potential.
Figures
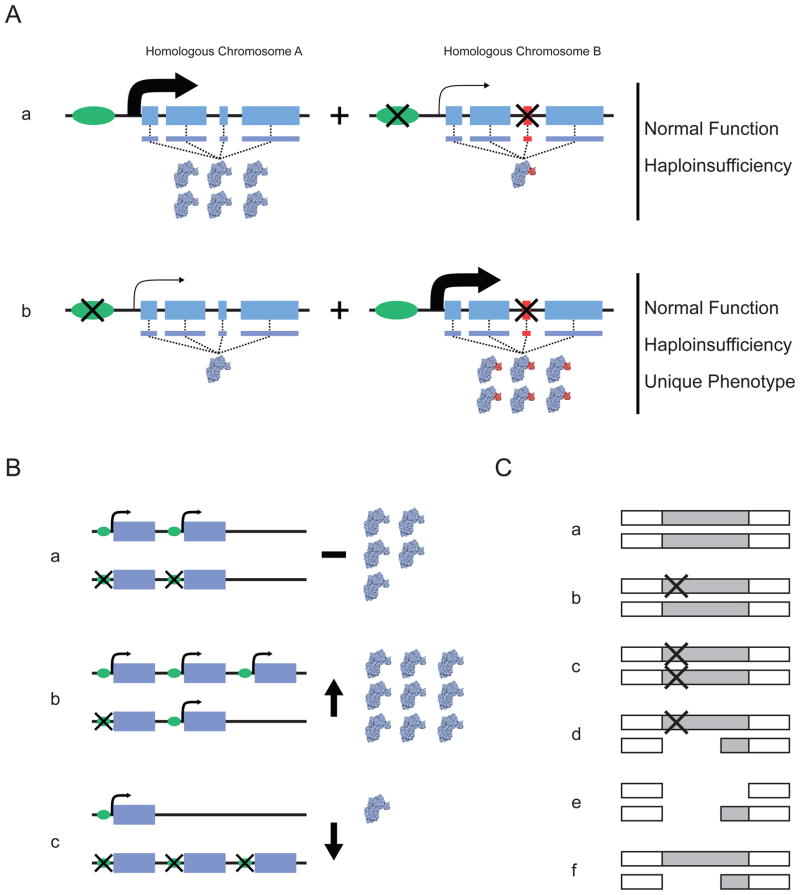
A | Distribution of variants that affect regulation and protein function, showing the two homologous gene segments in a single diploid individual. Aa | In this case, the leftmost homologue does not contain variation that influences either the expression or the structure of the encoded protein. By contrast, the rightmost homologue contains sequence variation in the promoter that reduces overall expression of the gene and exonic sequence variation that upsets the amino-acid sequence of the encoded protein. Ab | Here, the variants in the promoter and exonic sequence are distributed between different homologues. The combination of these homologues in a single individual can lead to haploinsufficiency if the homologue that does not have a functional variant cannot compensate for the affected homologue. If it can compensate, the overall functioning of the gene could be normal, owing to both the downregulation of the aberrant protein and the normal expression of the wild-type protein. B | Potential functional effects of haplotypes involving structural variants. Scenarios are shown involving copy-number variants and point mutations in a diploid setting. The possibilities depicted in parts Bb and Bc reflect increased and decreased overall gene expression, respectively, relative to that in Ba. C | Unmasking of deleterious mutations through gene deletion. A genomic region is shown that harbours a gene that is often either partially or completely deleted and that also harbours functionally relevant point mutations. Ca | Neither homologous copy of the gene harbours a variant. Cb | One of the gene homologues carries a point mutation. Cc | Both gene homologues carry a point mutation. Cd | One of the gene homologues carries a deletion and the other carries a point mutation. Ce | Both of the gene homologues carry a deletion. Cf | One of the gene homologues carries a deletion. Each situation could produce a different phenotype; for example, in part Cd the deletion depicted could unmask the deleterious effect of the point mutation on the other chromosome.
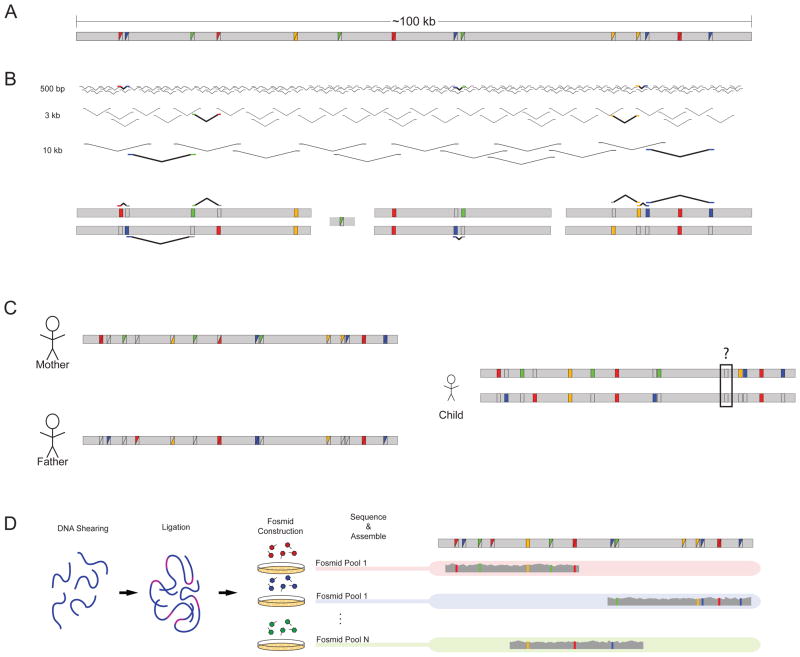
a | A hypothetical 100 kb stretch of sequence harbours multiple variants compared with the human reference, as designated by the coloured squares. Variants can be homozygous (solid coloured squares) or heterozygous (split coloured squares). b | Sequence reads from libraries of multiple insert sizes can be leveraged to link heterozygous sites together. Informative reads are highlighted and displayed a second time against the diploid reconstruction. The assembly consists of blocks of sequence with gaps arising when variants fall outside the distance of the insert sizes used for sequencing. c | Parental information allows for the separation of chromosomal variants except in instances in which both parents are heterozygous, as demonstrated by the black box in the child’s assembly. d | Laboratory-based methods such as the sequencing of fosmid pools allow for the separation of homologous chromosomes. DNA is sheared, ligated with fosmid vector sequence, packaged and transfected into the bacterium Escherichia coli. Pools of fosmid sequence — each containing only a small fraction of the total genome broken into ~40 kb segments — are sequenced independently. The sequenced libraries are then mapped and assembled for phase reconstruction.
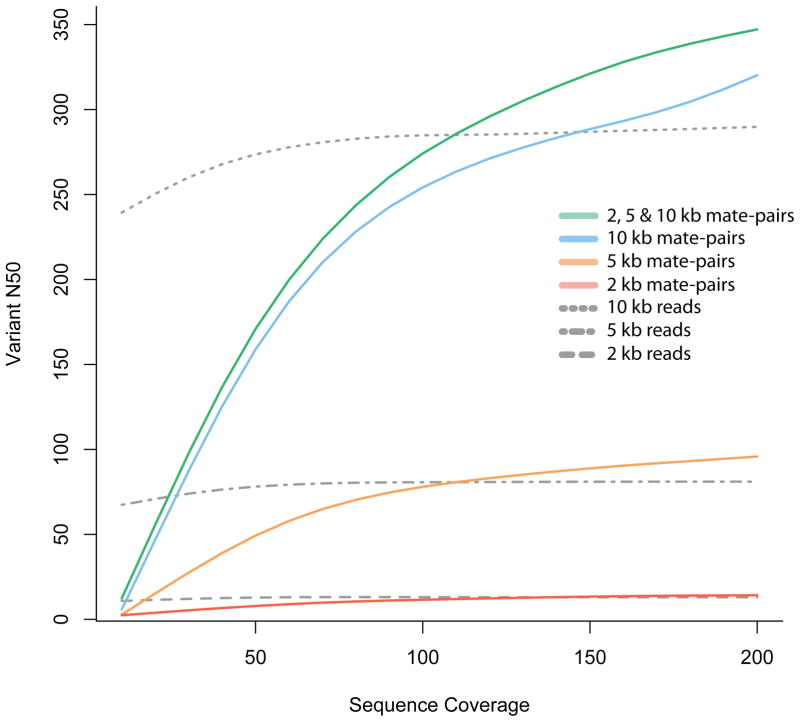
Simulated 100 bp mate-pair read coverage of various depths (sequence (fold) coverage, x-axis) for chromosome 1 of a Yoruban individual. All simulations were done using SNP calls (for chromosome 1) for the Yoruban individual NA19240, obtained from the 1000 Genomes project (released December 2008). Paired-end reads were simulated with the starting position of one read, chosen consistently at random on the chromosome, and the insert length sampled from a normal distribution with a given mean insert length (2, 5 or 10 kb) and standard deviation equal to 10% of the mean. For each simulation experiment, we constructed a graph with nodes corresponding to the heterozygous SNPs and edges corresponding to reads that cover multiple variants. The N50 was calculated using the number of variants in each connected component of this graph that correspond to the phased haplotype blocks. The vN50 is defined as the point at which half of the heterozygous loci of the chromosome are contained in contigs with the vN50 or greater number of variants. Mate-pair libraries outperform reads of the same length because the size distribution of the insert consists of lengths greater than 10 kb, allowing for longer connections than are possible with single reads alone.
References
-
- Lifton RP. Individual genomes on the horizon. N Engl J Med. 2010;362:1235–1236. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
