

Using transcription modules to identify expression clusters perturbed in Williams-Beuren syndrome
- PMID: 21304579
- PMCID: PMC3024257
- DOI: 10.1371/journal.pcbi.1001054
Using transcription modules to identify expression clusters perturbed in Williams-Beuren syndrome
Abstract
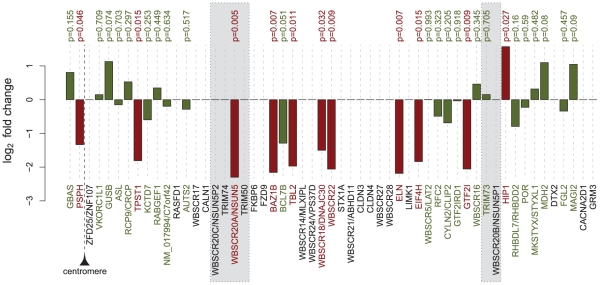
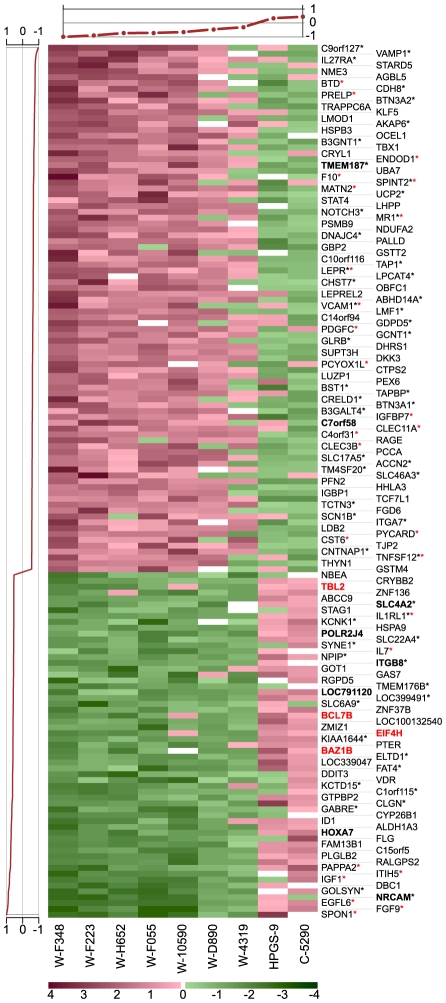
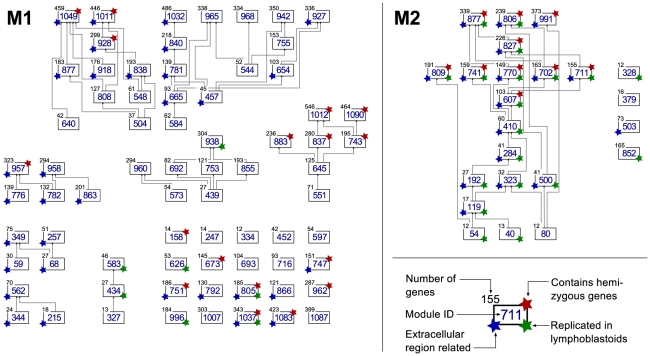
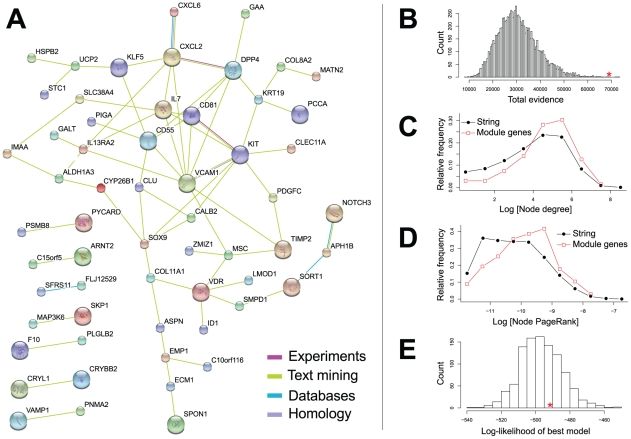
The genetic dissection of the phenotypes associated with Williams-Beuren Syndrome (WBS) is advancing thanks to the study of individuals carrying typical or atypical structural rearrangements, as well as in vitro and animal studies. However, little is known about the global dysregulations caused by the WBS deletion. We profiled the transcriptomes of skin fibroblasts from WBS patients and compared them to matched controls. We identified 868 differentially expressed genes that were significantly enriched in extracellular matrix genes, major histocompatibility complex (MHC) genes, as well as genes in which the products localize to the postsynaptic membrane. We then used public expression datasets from human fibroblasts to establish transcription modules, sets of genes coexpressed in this cell type. We identified those sets in which the average gene expression was altered in WBS samples. Dysregulated modules are often interconnected and share multiple common genes, suggesting that intricate regulatory networks connected by a few central genes are disturbed in WBS. This modular approach increases the power to identify pathways dysregulated in WBS patients, thus providing a testable set of additional candidates for genes and their interactions that modulate the WBS phenotypes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Attias J, Raveh E, Ben-Naftali N, Zarchi O, Gothelf D. Hyperactive auditory efferent system and lack of acoustic reflexes in Williams syndrome. J Basic Clin Physiol Pharmacol. 2008;19:193–207. - PubMed
-
- Cherniske EM, Carpenter TO, Klaiman C, Young E, Bregman J, et al. Multisystem study of 20 older adults with Williams syndrome. Am J Hum Genet. 2004;131:255–264. - PubMed
-
- Korenberg JR, Chen XN, Hirota H, Lai Z, Bellugi U, et al. VI. Genome structure and cognitive map of Williams syndrome. J Cognitive Neurosci. 2000;12(Suppl 1):89–107. - PubMed
-
- Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7:818–827. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
