

The architecture of gene regulatory variation across multiple human tissues: the MuTHER study
- PMID: 21304890
- PMCID: PMC3033383
- DOI: 10.1371/journal.pgen.1002003
The architecture of gene regulatory variation across multiple human tissues: the MuTHER study
Abstract
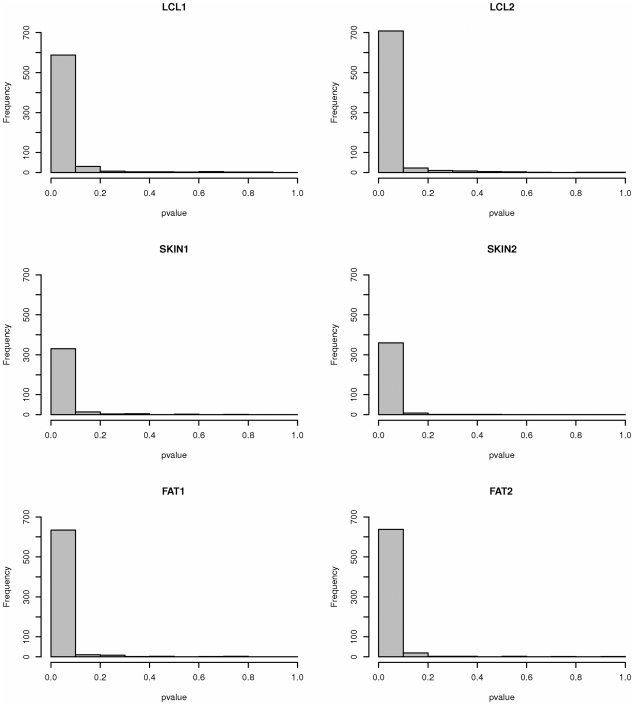
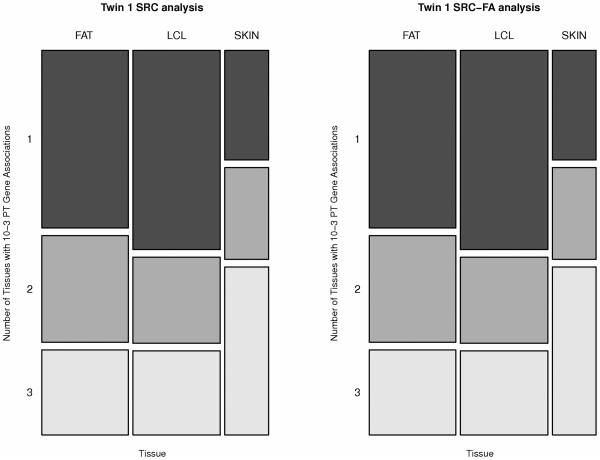
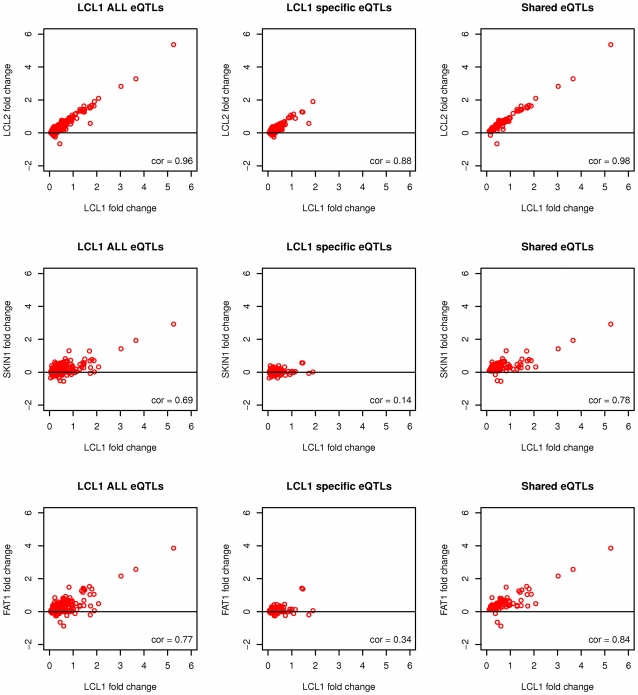
While there have been studies exploring regulatory variation in one or more tissues, the complexity of tissue-specificity in multiple primary tissues is not yet well understood. We explore in depth the role of cis-regulatory variation in three human tissues: lymphoblastoid cell lines (LCL), skin, and fat. The samples (156 LCL, 160 skin, 166 fat) were derived simultaneously from a subset of well-phenotyped healthy female twins of the MuTHER resource. We discover an abundance of cis-eQTLs in each tissue similar to previous estimates (858 or 4.7% of genes). In addition, we apply factor analysis (FA) to remove effects of latent variables, thus more than doubling the number of our discoveries (1,822 eQTL genes). The unique study design (Matched Co-Twin Analysis--MCTA) permits immediate replication of eQTLs using co-twins (93%-98%) and validation of the considerable gain in eQTL discovery after FA correction. We highlight the challenges of comparing eQTLs between tissues. After verifying previous significance threshold-based estimates of tissue-specificity, we show their limitations given their dependency on statistical power. We propose that continuous estimates of the proportion of tissue-shared signals and direct comparison of the magnitude of effect on the fold change in expression are essential properties that jointly provide a biologically realistic view of tissue-specificity. Under this framework we demonstrate that 30% of eQTLs are shared among the three tissues studied, while another 29% appear exclusively tissue-specific. However, even among the shared eQTLs, a substantial proportion (10%-20%) have significant differences in the magnitude of fold change between genotypic classes across tissues. Our results underline the need to account for the complexity of eQTL tissue-specificity in an effort to assess consequences of such variants for complex traits.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
