

Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development
- PMID: 21304941
- PMCID: PMC3033374
- DOI: 10.1371/journal.pgen.1001290
Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development
Abstract
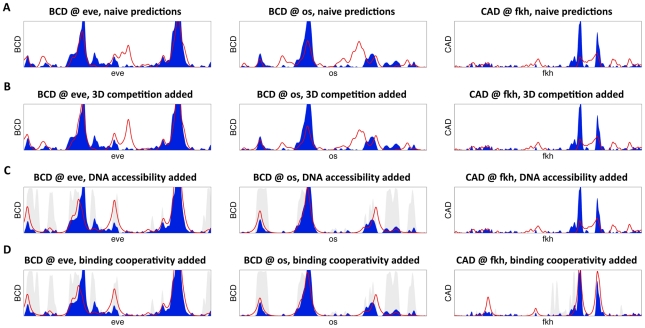
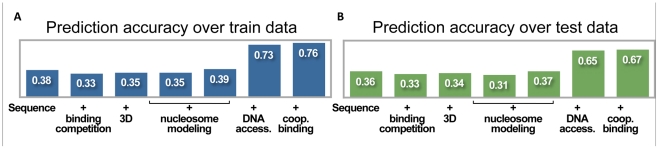
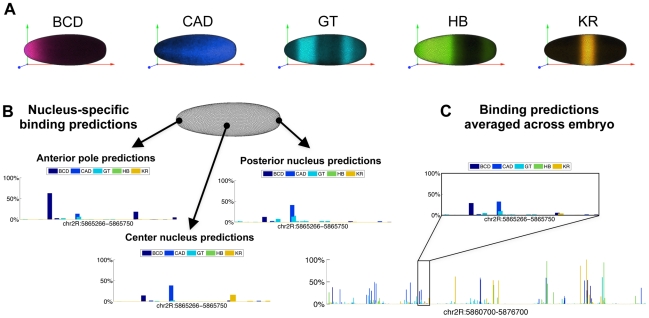
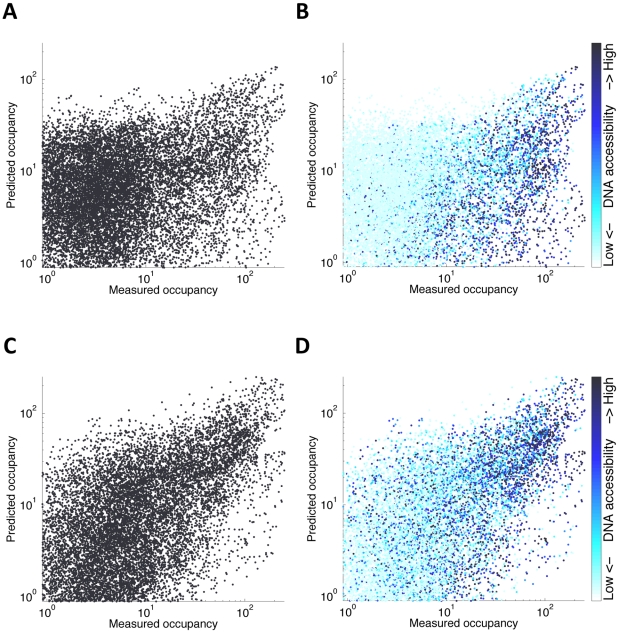
Transcription factors that drive complex patterns of gene expression during animal development bind to thousands of genomic regions, with quantitative differences in binding across bound regions mediating their activity. While we now have tools to characterize the DNA affinities of these proteins and to precisely measure their genome-wide distribution in vivo, our understanding of the forces that determine where, when, and to what extent they bind remains primitive. Here we use a thermodynamic model of transcription factor binding to evaluate the contribution of different biophysical forces to the binding of five regulators of early embryonic anterior-posterior patterning in Drosophila melanogaster. Predictions based on DNA sequence and in vitro protein-DNA affinities alone achieve a correlation of ∼0.4 with experimental measurements of in vivo binding. Incorporating cooperativity and competition among the five factors, and accounting for spatial patterning by modeling binding in every nucleus independently, had little effect on prediction accuracy. A major source of error was the prediction of binding events that do not occur in vivo, which we hypothesized reflected reduced accessibility of chromatin. To test this, we incorporated experimental measurements of genome-wide DNA accessibility into our model, effectively restricting predicted binding to regions of open chromatin. This dramatically improved our predictions to a correlation of 0.6-0.9 for various factors across known target genes. Finally, we used our model to quantify the roles of DNA sequence, accessibility, and binding competition and cooperativity. Our results show that, in regions of open chromatin, binding can be predicted almost exclusively by the sequence specificity of individual factors, with a minimal role for protein interactions. We suggest that a combination of experimentally determined chromatin accessibility data and simple computational models of transcription factor binding may be used to predict the binding landscape of any animal transcription factor with significant precision.
Conflict of interest statement
MBE is a co-founder and member of the Board of Directors of PLoS.
Figures
References
-
- Walter J, Dever CA, Biggin MD. Two homeo domain proteins bind with similar specificity to a wide range of DNA sites in Drosophila embryos. Genes Dev. 1994;8:1678–1692. - PubMed
-
- Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell. 2006;24:593–602. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
