

Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: a dual role for p53
- PMID: 21304979
- PMCID: PMC3031577
- DOI: 10.1371/journal.pone.0016523
Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: a dual role for p53
Abstract
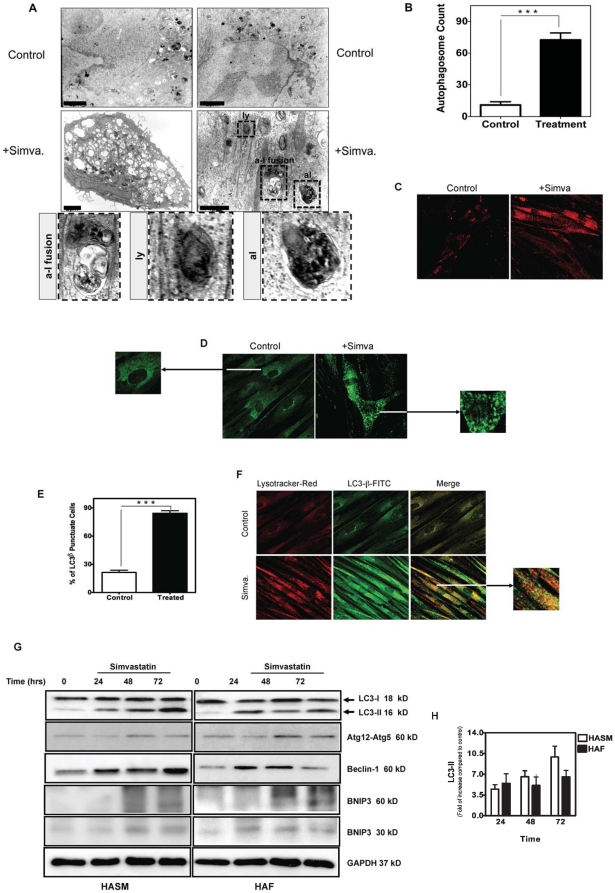
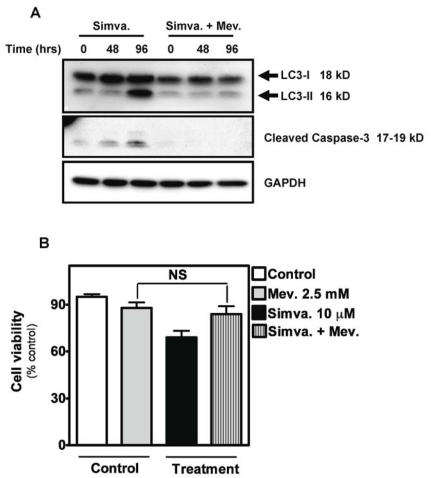
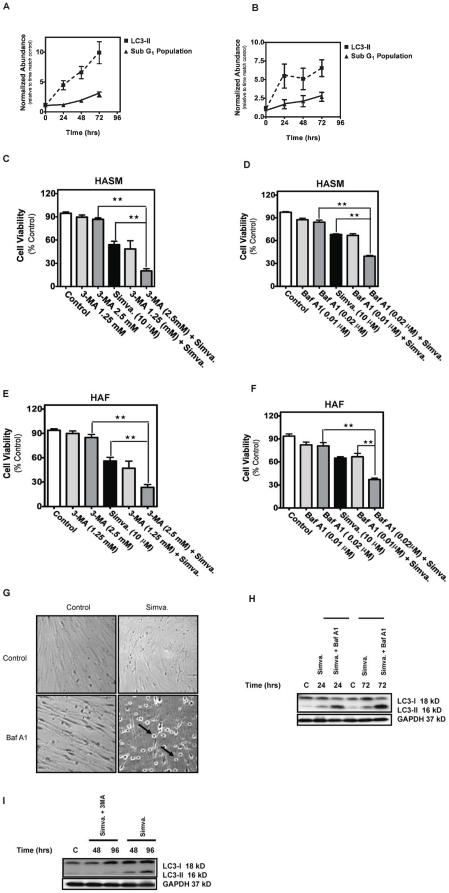
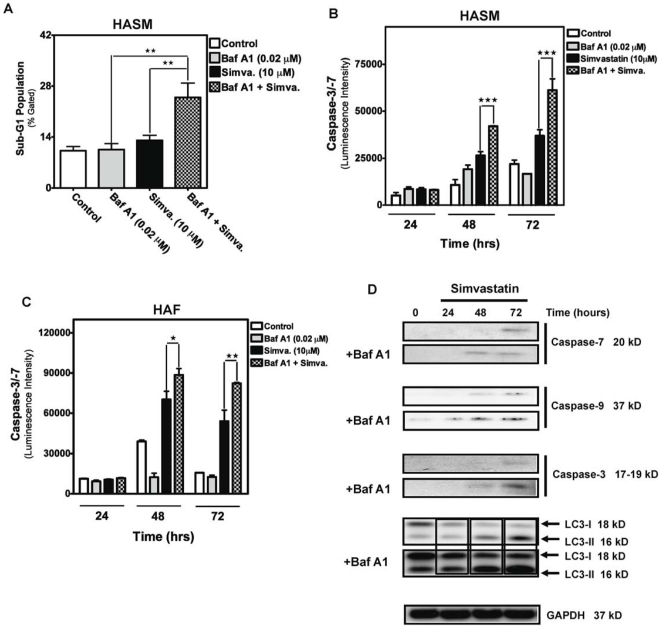
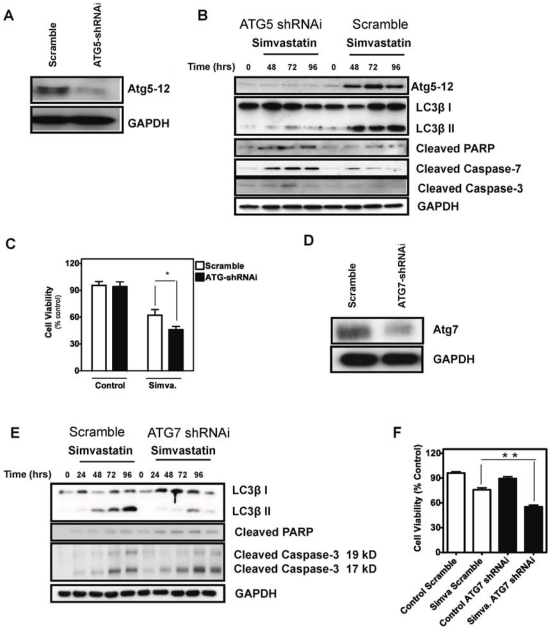
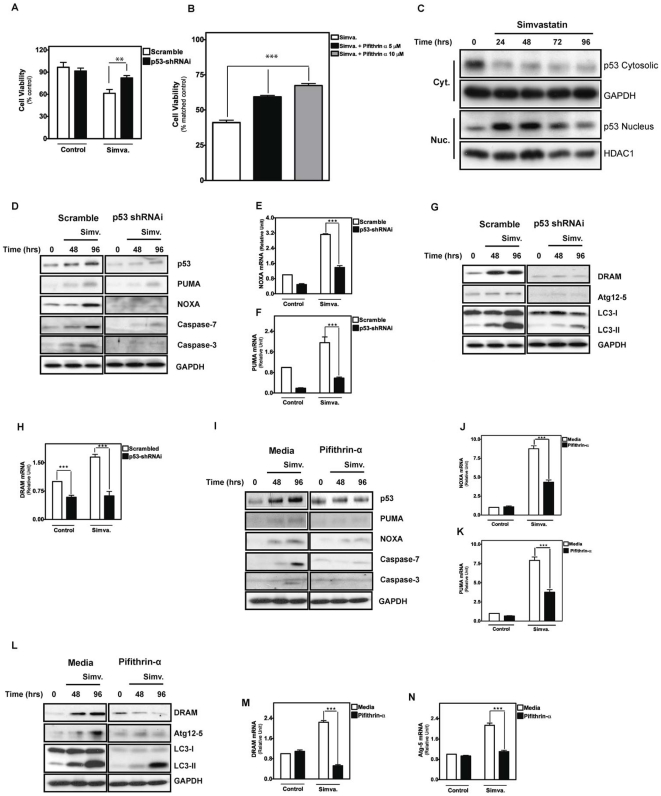
Statins inhibit the proximal steps of cholesterol biosynthesis, and are linked to health benefits in various conditions, including cancer and lung disease. We have previously investigated apoptotic pathways triggered by statins in airway mesenchymal cells, and identified reduced prenylation of small GTPases as a primary effector mechanism leading to p53-mediated cell death. Here, we extend our studies of statin-induced cell death by assessing endpoints of both apoptosis and autophagy, and investigating their interplay and coincident regulation. Using primary cultured human airway smooth muscle (HASM) and human airway fibroblasts (HAF), autophagy, and autophagosome formation and flux were assessed by transmission electron microscopy, cytochemistry (lysosome number and co-localization with LC3) and immunoblotting (LC3 lipidation and Atg12-5 complex formation). Chemical inhibition of autophagy increased simvastatin-induced caspase activation and cell death. Similarly, Atg5 silencing with shRNA, thus preventing Atg5-12 complex formation, increased pro-apoptotic effects of simvastatin. Simvastatin concomitantly increased p53-dependent expression of p53 up-regulated modulator of apoptosis (PUMA), NOXA, and damage-regulated autophagy modulator (DRAM). Notably both mevalonate cascade inhibition-induced autophagy and apoptosis were p53 dependent: simvastatin increased nuclear p53 accumulation, and both cyclic pifithrin-α and p53 shRNAi partially inhibited NOXA, PUMA expression and caspase-3/7 cleavage (apoptosis) and DRAM expression, Atg5-12 complex formation, LC3 lipidation, and autophagosome formation (autophagy). Furthermore, the autophagy response is induced rapidly, significantly delaying apoptosis, suggesting the existence of a temporally coordinated p53 regulation network. These findings are relevant for the development of statin-based therapeutic approaches in obstructive airway disease.
Conflict of interest statement
Figures
Similar articles
-
Airway mesenchymal cell death by mevalonate cascade inhibition: integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins.Biochim Biophys Acta. 2014 Jul;1843(7):1259-71. doi: 10.1016/j.bbamcr.2014.03.006. Epub 2014 Mar 15. Biochim Biophys Acta. 2014. PMID: 24637330
-
Statin-triggered cell death in primary human lung mesenchymal cells involves p53-PUMA and release of Smac and Omi but not cytochrome c.Biochim Biophys Acta. 2010 Apr;1803(4):452-67. doi: 10.1016/j.bbamcr.2009.12.005. Epub 2010 Jan 4. Biochim Biophys Acta. 2010. PMID: 20045437
-
Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle.Am J Physiol Lung Cell Mol Physiol. 2012 Feb 15;302(4):L420-8. doi: 10.1152/ajplung.00312.2011. Epub 2011 Dec 9. Am J Physiol Lung Cell Mol Physiol. 2012. PMID: 22160308
-
Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy.Autophagy. 2010 Jan;6(1):153-4. doi: 10.4161/auto.6.1.10537. Epub 2010 Jan 5. Autophagy. 2010. PMID: 19949306 Review.
-
Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link.Int J Mol Sci. 2015 Jul 15;16(7):16067-84. doi: 10.3390/ijms160716067. Int J Mol Sci. 2015. PMID: 26184189 Free PMC article. Review.
Cited by
-
Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes.Cell Physiol Biochem. 2013;31(6):925-37. doi: 10.1159/000350111. Epub 2013 Jun 18. Cell Physiol Biochem. 2013. PMID: 23817226 Free PMC article.
-
Simvastatin Induces Apoptosis in Medulloblastoma Brain Tumor Cells via Mevalonate Cascade Prenylation Substrates.Cancers (Basel). 2019 Jul 17;11(7):994. doi: 10.3390/cancers11070994. Cancers (Basel). 2019. PMID: 31319483 Free PMC article.
-
Give Me a Fork: Can Autophagy Research Solve the Riddle of Airway Remodeling in Asthma?Am J Respir Cell Mol Biol. 2019 May;60(5):494-496. doi: 10.1165/rcmb.2018-0353ED. Am J Respir Cell Mol Biol. 2019. PMID: 30423254 Free PMC article. No abstract available.
-
BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis.Life (Basel). 2022 Feb 9;12(2):256. doi: 10.3390/life12020256. Life (Basel). 2022. PMID: 35207544 Free PMC article. Review.
-
Statins and asthma: where we stand, and the next critical steps in research.Curr Med Res Opin. 2014 Jun;30(6):1051-4. doi: 10.1185/03007995.2014.881794. Epub 2014 Jan 30. Curr Med Res Opin. 2014. PMID: 24450485 Free PMC article.
References
-
- Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510. - PubMed
-
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. - PubMed
-
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. - PubMed
-
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–458. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
