

EGF signaling regulates the proliferation of intestinal stem cells in Drosophila
- PMID: 21307097
- PMCID: PMC3042864
- DOI: 10.1242/dev.056671
EGF signaling regulates the proliferation of intestinal stem cells in Drosophila
Abstract
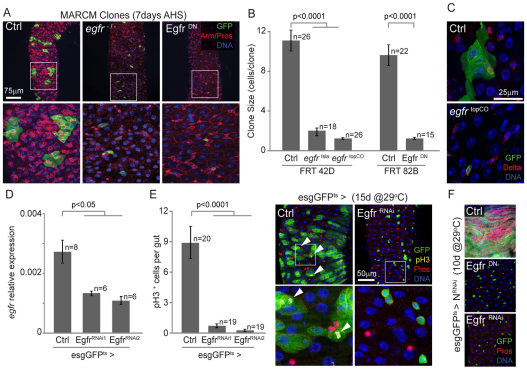
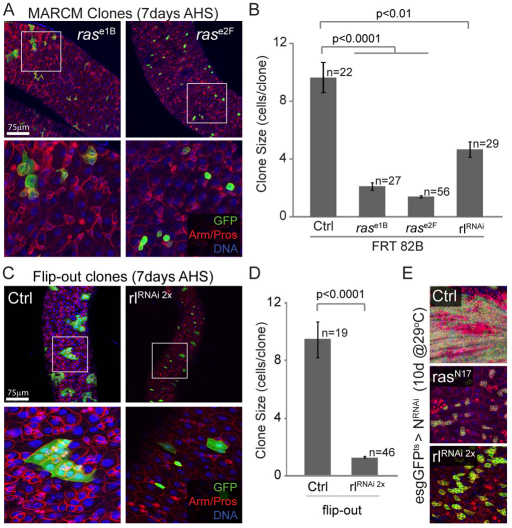
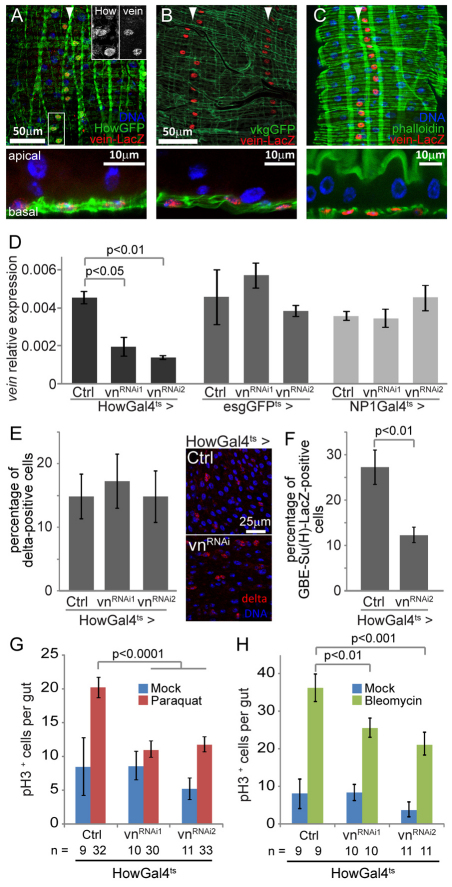
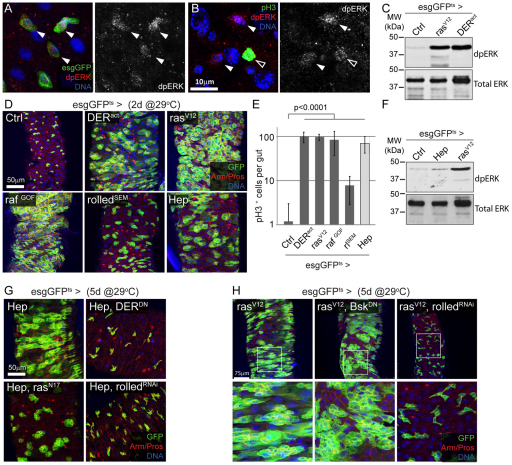
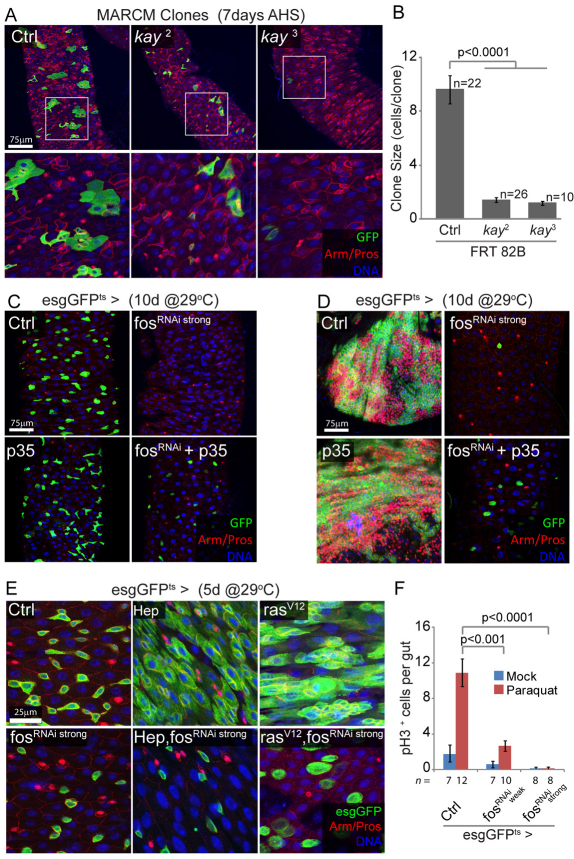
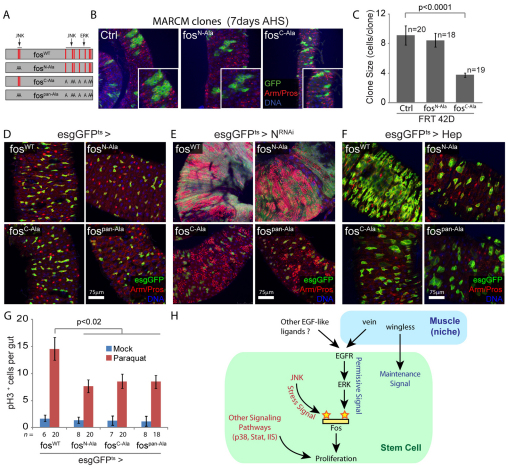
Precise control of somatic stem cell proliferation is crucial to ensure maintenance of tissue homeostasis in high-turnover tissues. In Drosophila, intestinal stem cells (ISCs) are essential for homeostatic turnover of the intestinal epithelium and ensure epithelial regeneration after tissue damage. To accommodate these functions, ISC proliferation is regulated dynamically by various growth factors and stress signaling pathways. How these signals are integrated is poorly understood. Here, we show that EGF receptor signaling is required to maintain the proliferative capacity of ISCs. The EGF ligand Vein is expressed in the muscle surrounding the intestinal epithelium, providing a permissive signal for ISC proliferation. We find that the AP-1 transcription factor FOS serves as a convergence point for this signal and for the Jun N-terminal kinase (JNK) pathway, which promotes ISC proliferation in response to stress. Our results support the notion that the visceral muscle serves as a functional 'niche' for ISCs, and identify FOS as a central integrator of a niche-derived permissive signal with stress-induced instructive signals, adjusting ISC proliferation to environmental conditions.
Figures
References
-
- Baker N. E., Rubin G. M. (1989). Effect on eye development of dominant mutations in Drosophila homologue of the EGF receptor. Nature 340, 150-153 - PubMed
-
- Barker N., Ridgway R. A., van Es J. H., van de Wetering M., Begthel H., van den Born M., Danenberg E., Clarke A. R., Sansom O. J., Clevers H. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608-611 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
