

Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo
- PMID: 21307261
- PMCID: PMC3092483
- DOI: 10.1523/JNEUROSCI.3153-09.2011
Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo
Abstract
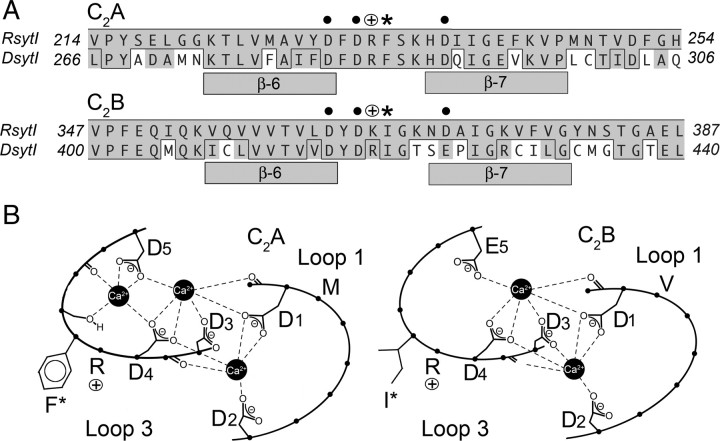
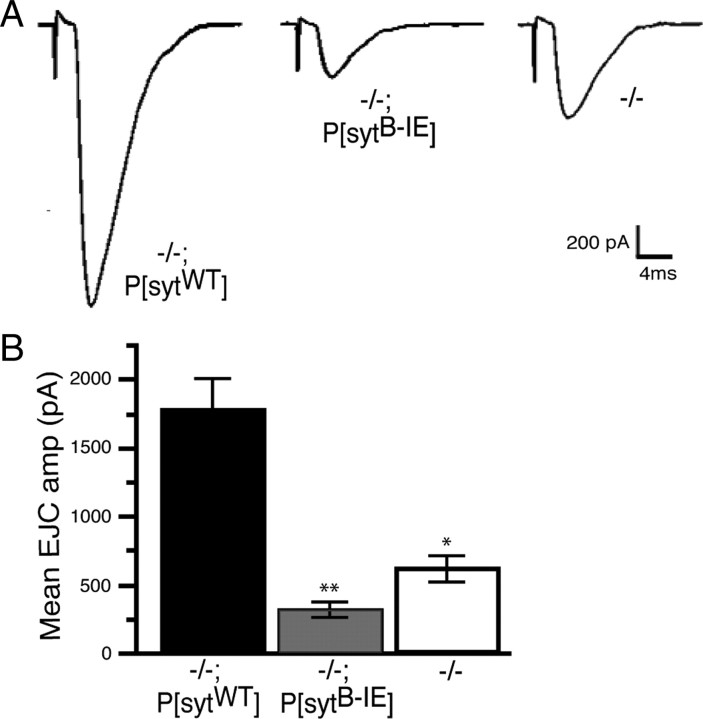
The vesicle protein synaptotagmin I is the Ca(2+) sensor that triggers fast, synchronous release of neurotransmitter. Specifically, Ca(2+) binding by the C(2)B domain of synaptotagmin is required at intact synapses, yet the mechanism whereby Ca(2+) binding results in vesicle fusion remains controversial. Ca(2+)-dependent interactions between synaptotagmin and SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment receptor) complexes and/or anionic membranes are possible effector interactions. However, no effector-interaction mutations to date impact synaptic transmission as severely as mutation of the C(2)B Ca(2+)-binding motif, suggesting that these interactions are facilitatory rather than essential. Here we use Drosophila to show the functional role of a highly conserved, hydrophobic residue located at the tip of each of the two Ca(2+)-binding pockets of synaptotagmin. Mutation of this residue in the C(2)A domain (F286) resulted in a ∼50% decrease in evoked transmitter release at an intact synapse, again indicative of a facilitatory role. Mutation of this hydrophobic residue in the C(2)B domain (I420), on the other hand, blocked all locomotion, was embryonic lethal even in syt I heterozygotes, and resulted in less evoked transmitter release than that in syt(null) mutants, which is more severe than the phenotype of C(2)B Ca(2+)-binding mutants. Thus, mutation of a single, C(2)B hydrophobic residue required for Ca(2+)-dependent penetration of anionic membranes results in the most severe disruption of synaptotagmin function in vivo to date. Our results provide direct support for the hypothesis that plasma membrane penetration, specifically by the C(2)B domain of synaptotagmin, is the critical effector interaction for coupling Ca(2+) binding with vesicle fusion.
Figures





References
-
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. - PubMed
-
- Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem. 1995;270:23667–23671. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous