

Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire
- PMID: 21310087
- PMCID: PMC3045962
- DOI: 10.1186/1471-2164-12-106
Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire
Abstract
Background: Recent advances in massively parallel sequencing have increased the depth at which T cell receptor (TCR) repertoires can be probed by >3log10, allowing for saturation sequencing of immune repertoires. The resolution of this sequencing is dependent on its accuracy, and direct assessments of the errors formed during high throughput repertoire analyses are limited.
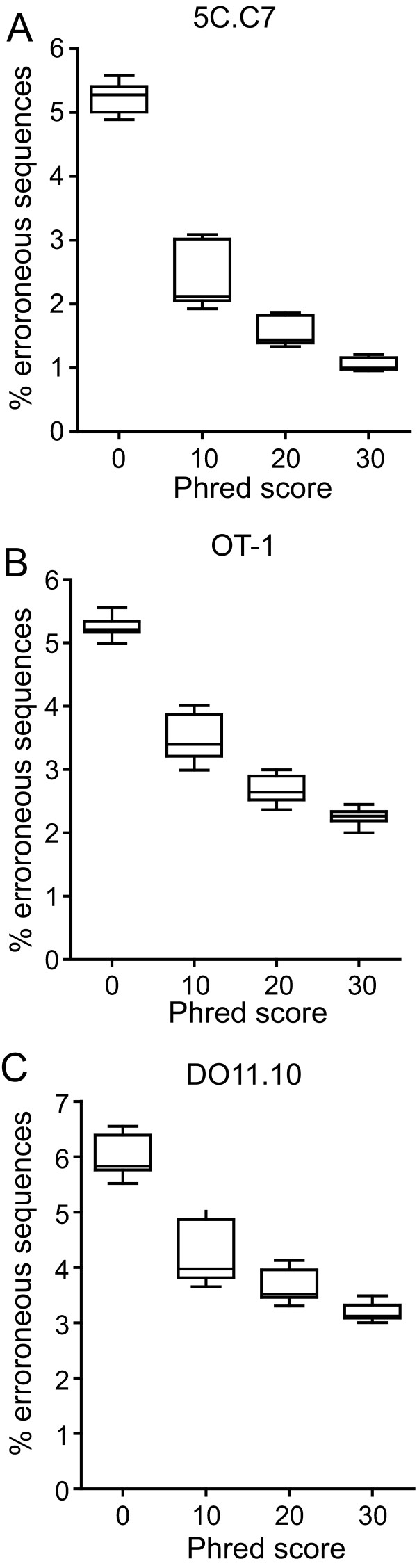
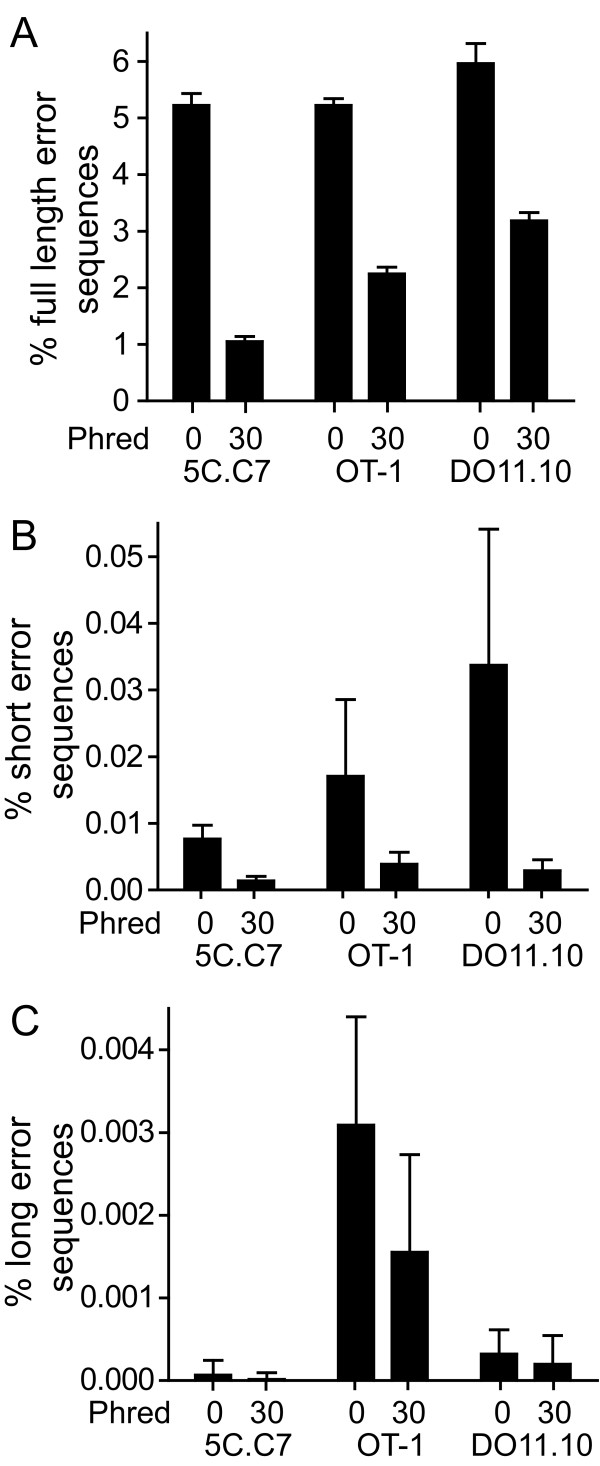
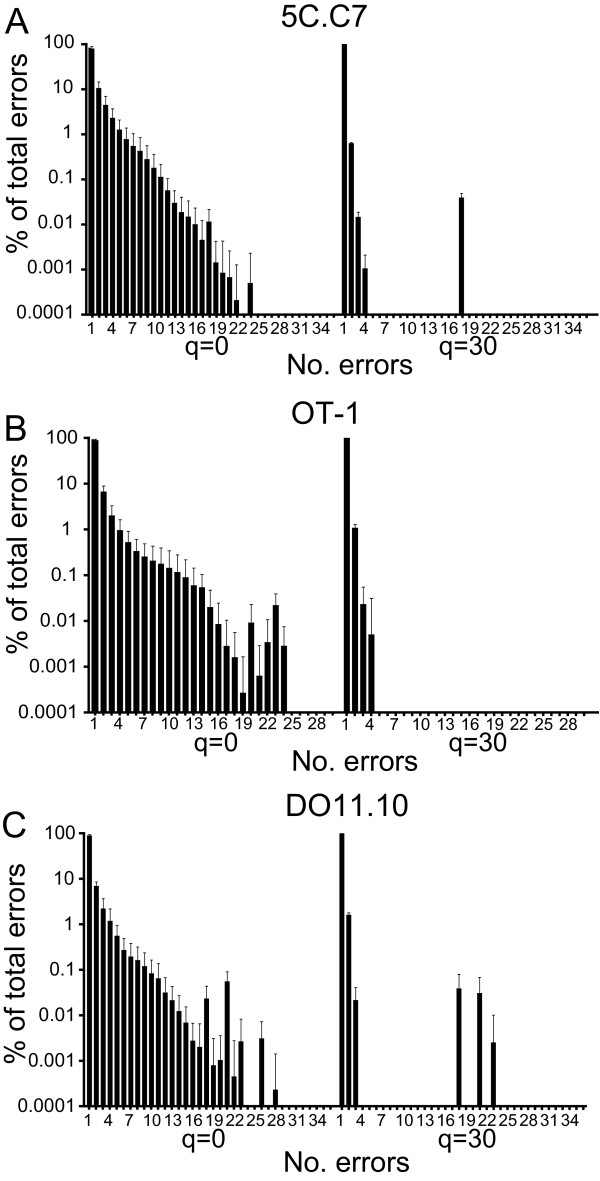
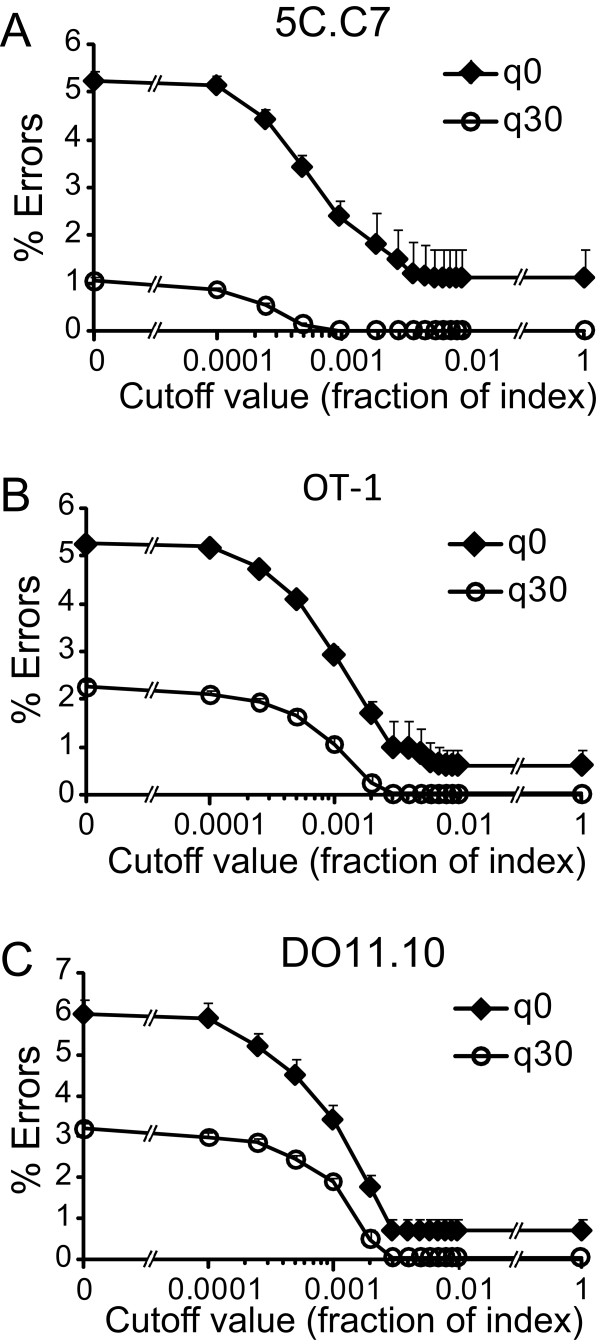
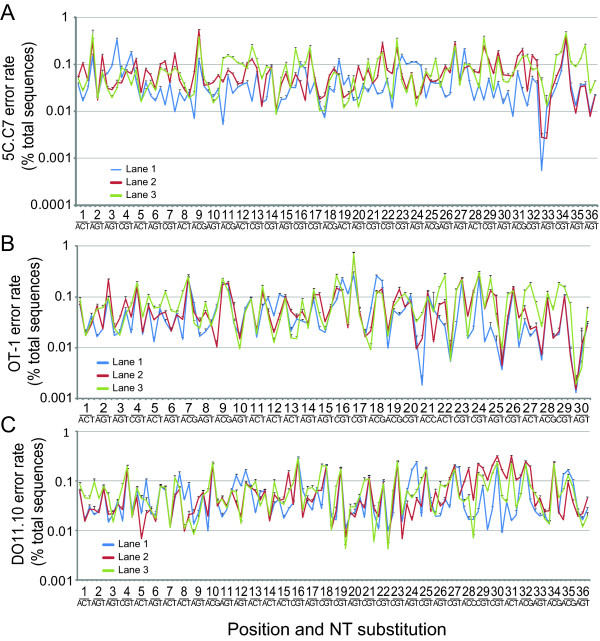
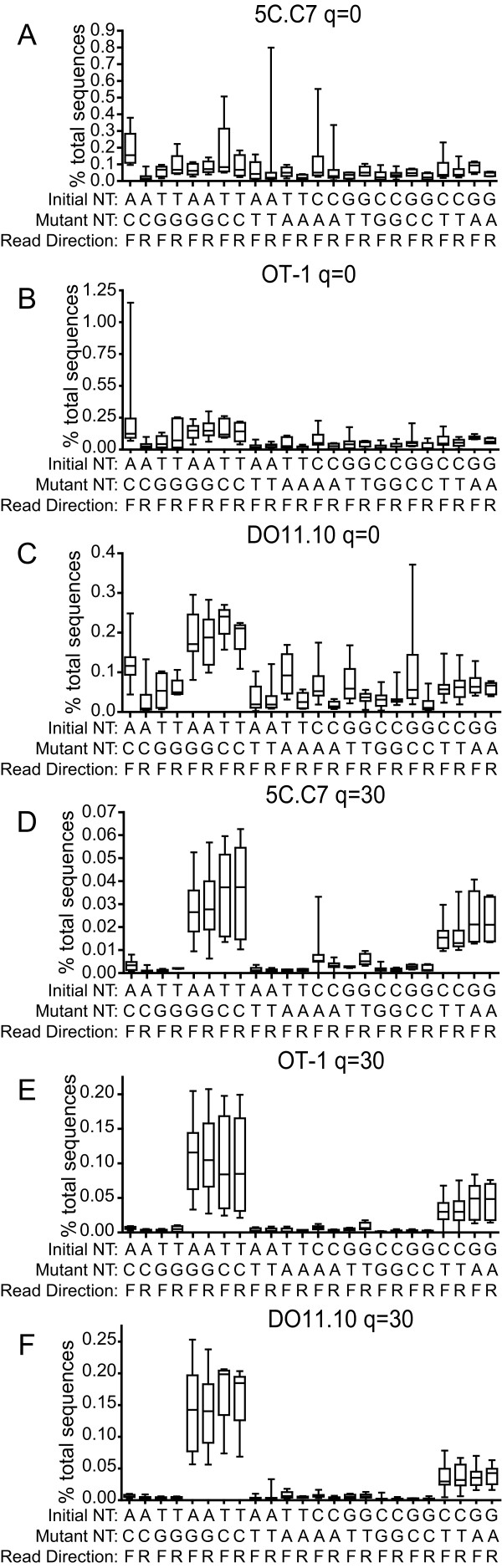
Results: We analyzed 3 monoclonal TCR from TCR transgenic, Rag-/- mice using Illumina® sequencing. A total of 27 sequencing reactions were performed for each TCR using a trifurcating design in which samples were divided into 3 at significant processing junctures. More than 20 million complementarity determining region (CDR) 3 sequences were analyzed. Filtering for lower quality sequences diminished but did not eliminate sequence errors, which occurred within 1-6% of sequences. Erroneous sequences were pre-dominantly of correct length and contained single nucleotide substitutions. Rates of specific substitutions varied dramatically in a position-dependent manner. Four substitutions, all purine-pyrimidine transversions, predominated. Solid phase amplification and sequencing rather than liquid sample amplification and preparation appeared to be the primary sources of error. Analysis of polyclonal repertoires demonstrated the impact of error accumulation on data parameters.
Conclusions: Caution is needed in interpreting repertoire data due to potential contamination with mis-sequence reads. However, a high association of errors with phred score, high relatedness of erroneous sequences with the parental sequence, dominance of specific nt substitutions, and skewed ratio of forward to reverse reads among erroneous sequences indicate approaches to filter erroneous sequences from repertoire data sets.
Figures



References
-
- Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. 2000;164:5782–5787. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
