

Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency
- PMID: 21310276
- PMCID: PMC3035707
- DOI: 10.1016/j.ajhg.2011.01.004
Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency
Abstract
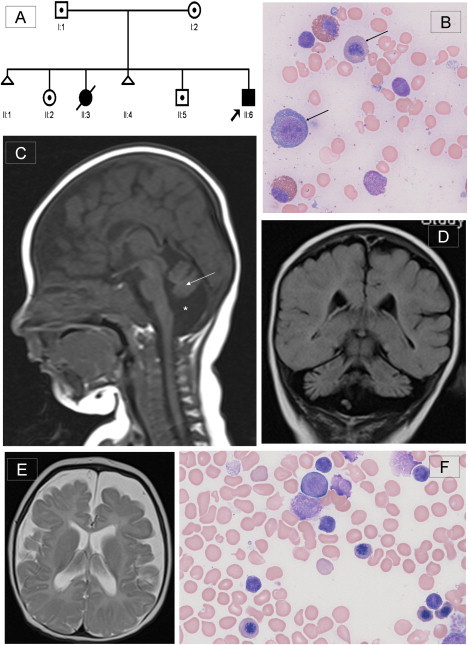
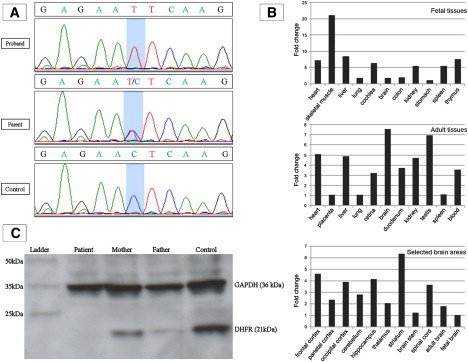
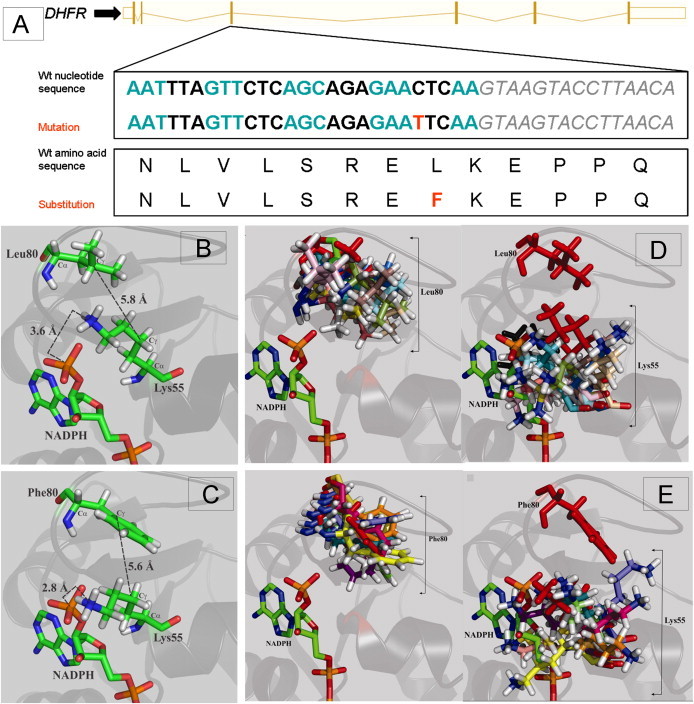
Dihydrofolate reductase (DHFR) is a critical enzyme in folate metabolism and an important target of antineoplastic, antimicrobial, and antiinflammatory drugs. We describe three individuals from two families with a recessive inborn error of metabolism, characterized by megaloblastic anemia and/or pancytopenia, severe cerebral folate deficiency, and cerebral tetrahydrobiopterin deficiency due to a germline missense mutation in DHFR, resulting in profound enzyme deficiency. We show that cerebral folate levels, anemia, and pancytopenia of DHFR deficiency can be corrected by treatment with folinic acid. The characterization of this disorder provides evidence for the link between DHFR and metabolism of cerebral tetrahydrobiopterin, which is required for the formation of dopamine, serotonin, and norepinephrine and for the hydroxylation of aromatic amino acids. Moreover, this relationship provides insight into the role of folates in neurological conditions, including depression, Alzheimer disease, and Parkinson disease.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Djukic A. Folate-Responsive Neurologic Diseases. Pediatr. Neurol. 2007;37:387–397. - PubMed
-
- Steinfeld R., Grapp M., Kraetzner R., Dreha-Kulaczewski S., Helms G., Dechent P., Wevers R., Grosso S., Gärtner J. Folate Receptor Alpha Defect Causes Cerebral Folate Transport Deficiency: A Treatable Neurodegenerative Disorder Associated with Disturbed Myelin Metabolism. Am. J. Hum. Genet. 2009;85:354–363. - PMC - PubMed
-
- Ramaekers V.T., Rothenberg S.P., Sequeira J.M., Opladen T., Blau N., Quadros E.V., Selhub J. Autoantibodies to Folate Receptors in the Cerebral Folate Deficiency Syndrome. N. Engl. J. Med. 2005;352:1985–1991. - PubMed
-
- Hyland K., Surtees R.A.H., Heales S.J.R., Bowron A., Howells D.W., Smith I. Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr. Res. 1993;34:10–14. - PubMed
-
- Qiu A., Jansen M., Sakaris A., Min S.H., Chattopadhyay S., Tsai E., Sandoval C., Zhao R., Akabas M.H., Goldman I.D. Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell. 2006;127:917–928. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
