

Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure
- PMID: 21311045
- PMCID: PMC3785241
- DOI: 10.1161/CIRCRESAHA.110.232306
Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure
Abstract
Rationale: Mitochondrial dysfunction has been implicated in several cardiovascular diseases; however, the roles of mitochondrial oxidative stress and DNA damage in hypertensive cardiomyopathy are not well understood.
Objective: We evaluated the contribution of mitochondrial reactive oxygen species (ROS) to cardiac hypertrophy and failure by using genetic mouse models overexpressing catalase targeted to mitochondria and to peroxisomes.
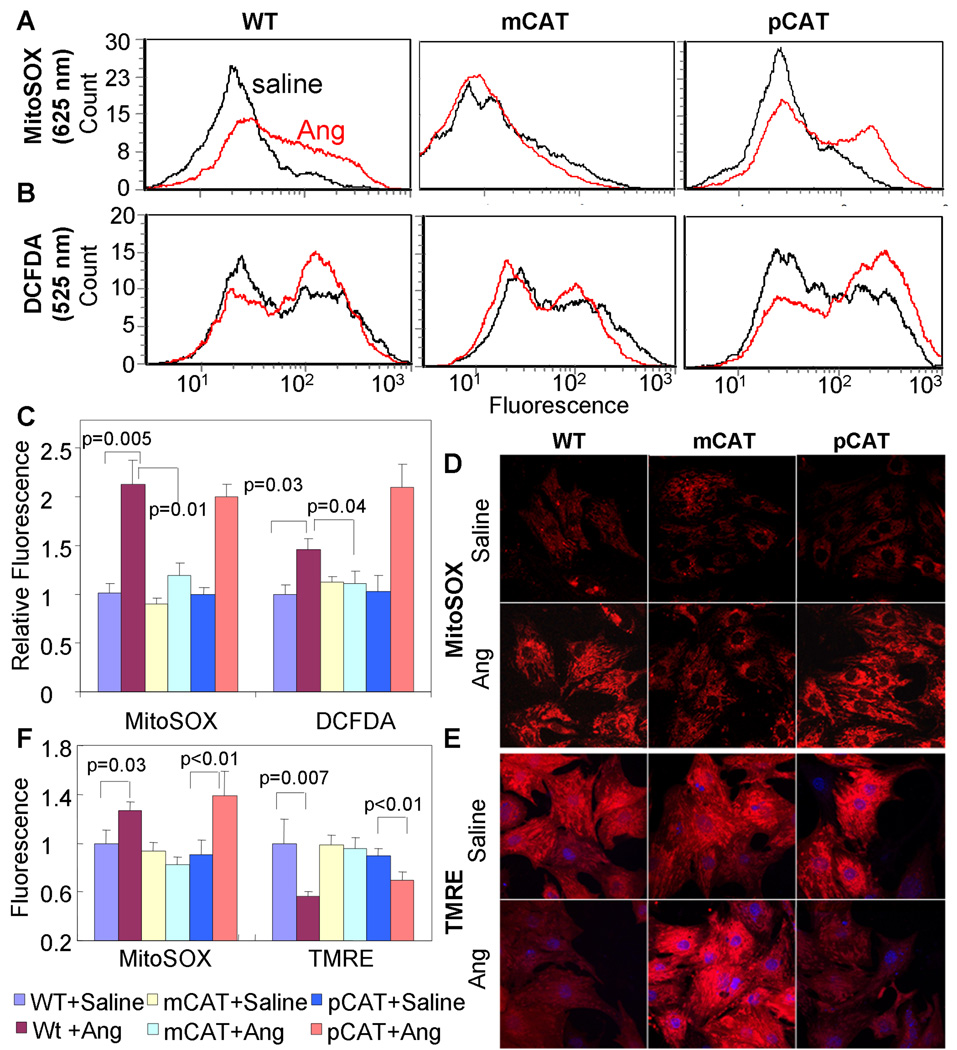
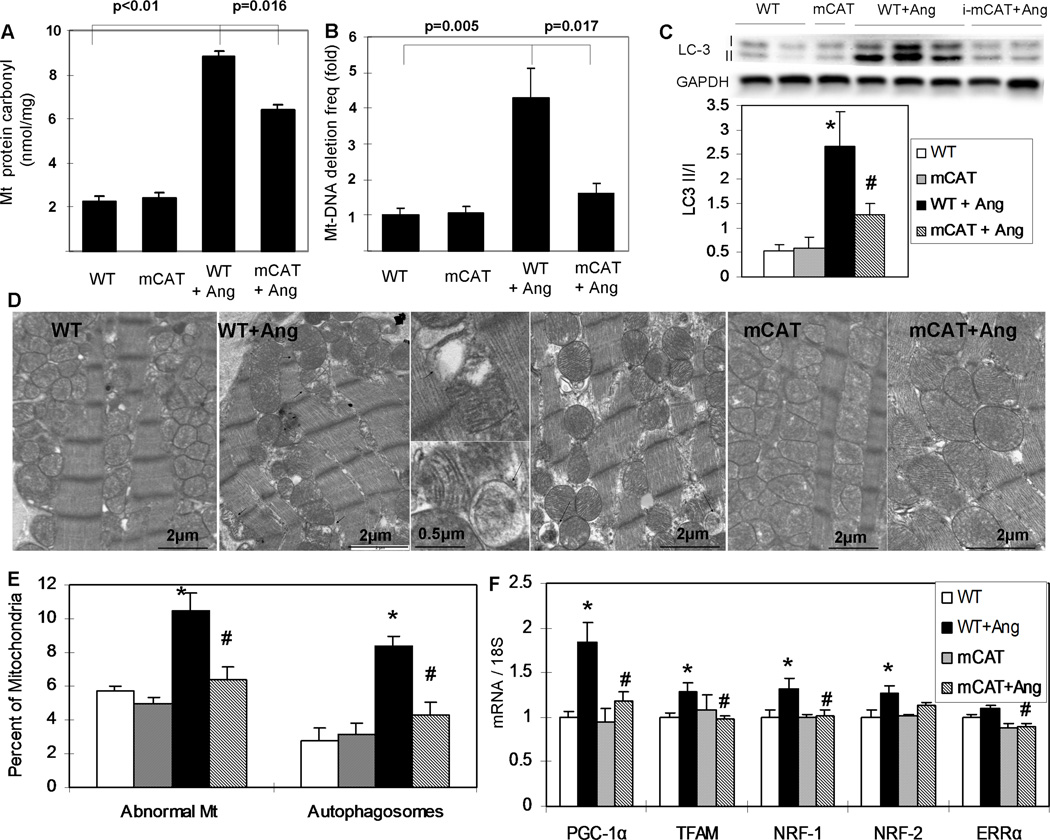
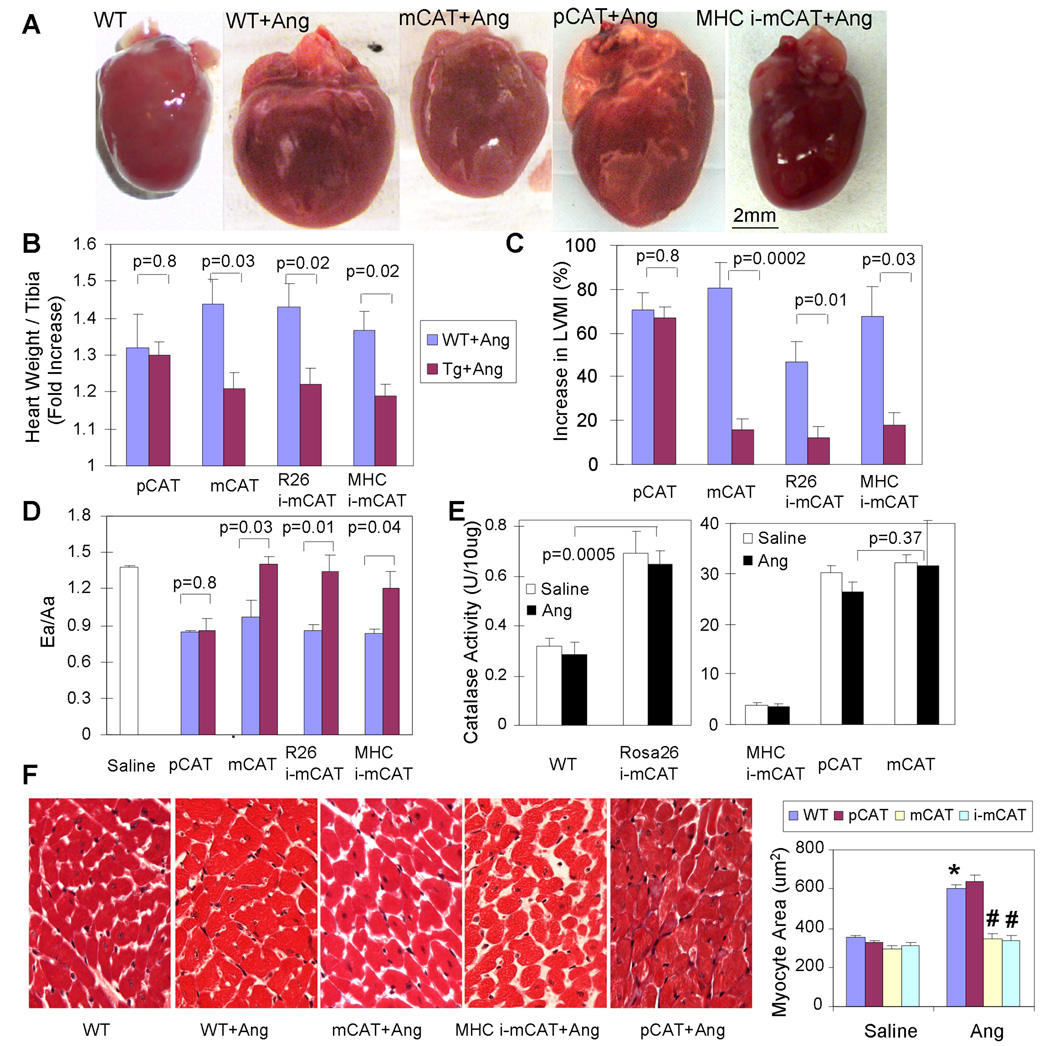
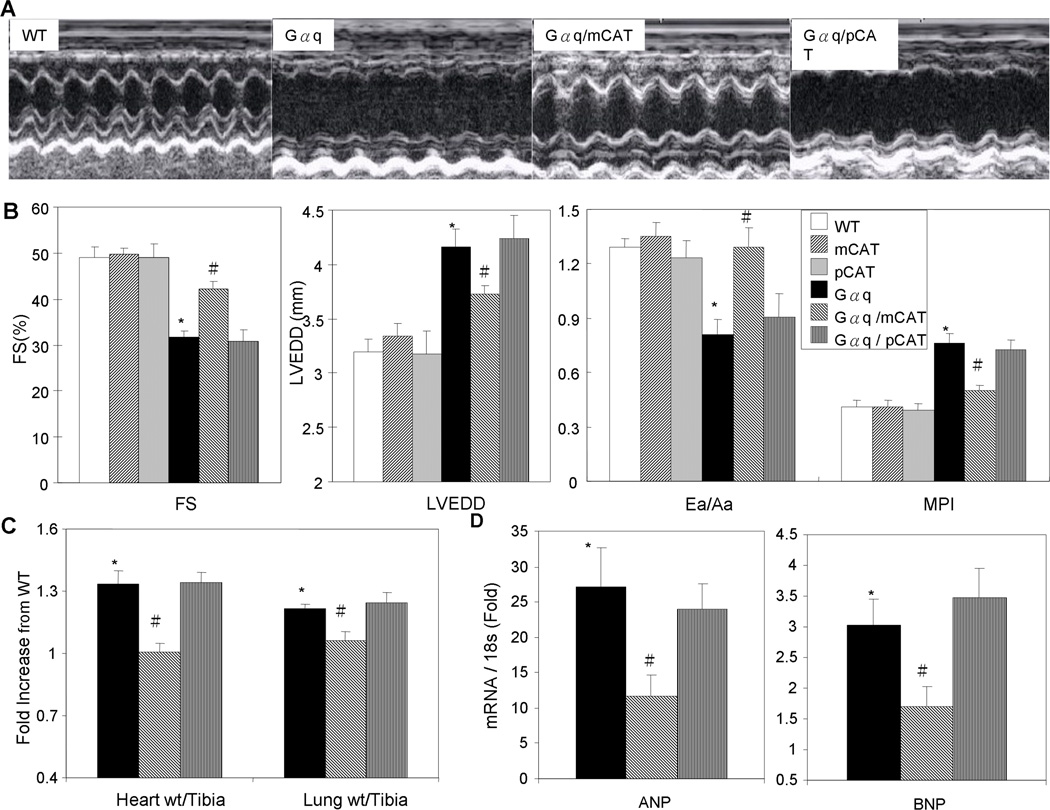
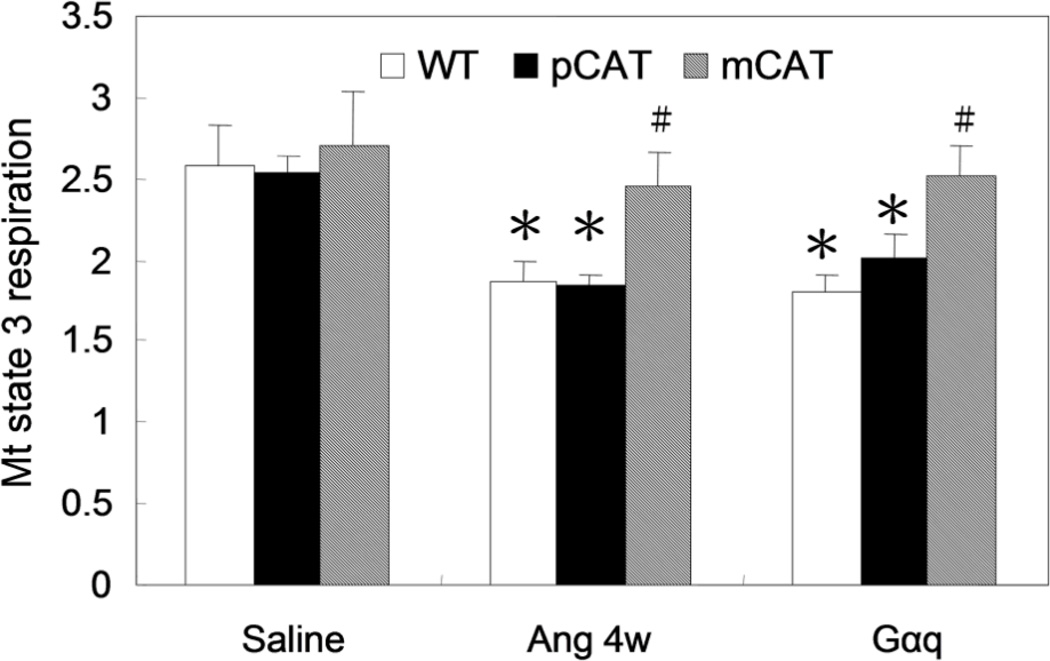
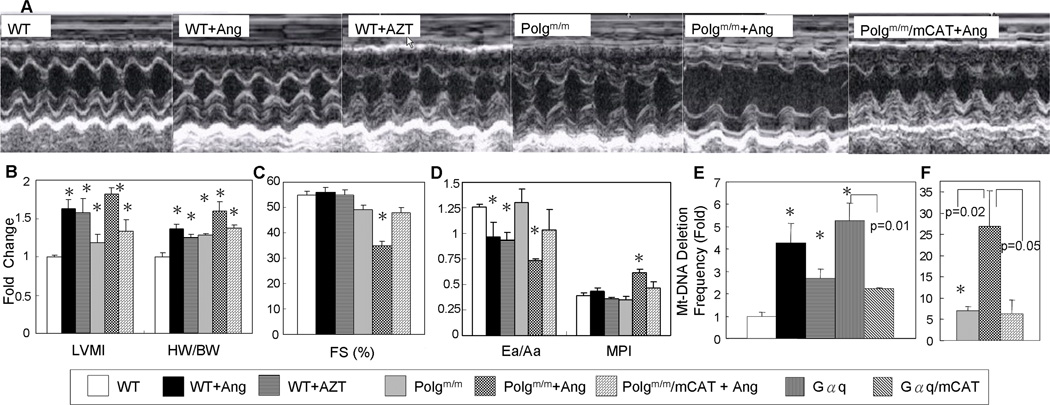
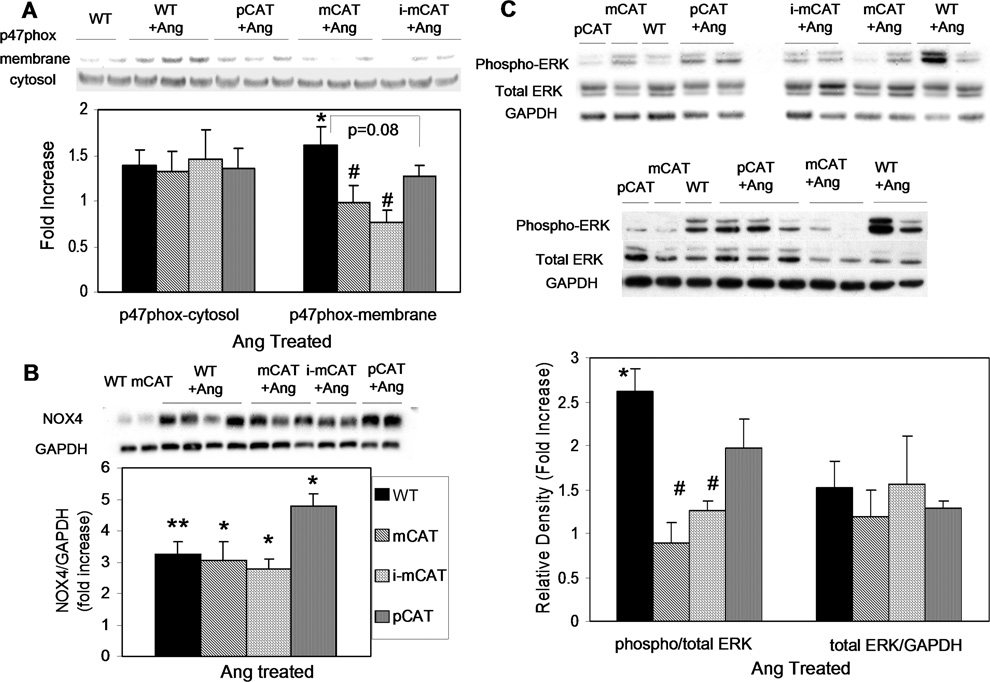
Methods and results: Angiotensin II increases mitochondrial ROS in cardiomyocytes, concomitant with increased mitochondrial protein carbonyls, mitochondrial DNA deletions, increased autophagy and signaling for mitochondrial biogenesis in hearts of angiotensin II-treated mice. The causal role of mitochondrial ROS in angiotensin II-induced cardiomyopathy is shown by the observation that mice that overexpress catalase targeted to mitochondria, but not mice that overexpress wild-type peroxisomal catalase, are resistant to cardiac hypertrophy, fibrosis and mitochondrial damage induced by angiotensin II, as well as heart failure induced by overexpression of Gαq. Furthermore, primary damage to mitochondrial DNA, induced by zidovudine administration or homozygous mutation of mitochondrial polymerase γ, is also shown to contribute directly to the development of cardiac hypertrophy, fibrosis and failure.
Conclusions: These data indicate the critical role of mitochondrial ROS in cardiac hypertrophy and failure and support the potential use of mitochondrial-targeted antioxidants for prevention and treatment of hypertensive cardiomyopathy.
Conflict of interest statement
Figures
References
-
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. - PubMed
-
- Gardin JM, Lauer MS. Left ventricular hypertrophy: the next treatable, silent killer? Jama. 2004;292:2396–2398. - PubMed
-
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. - PubMed
-
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. - PubMed
-
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45:438–444. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
