

Huntington's disease: can mice lead the way to treatment?
- PMID: 21315254
- PMCID: PMC4685469
- DOI: 10.1016/j.neuron.2010.12.035
Huntington's disease: can mice lead the way to treatment?
Erratum in
- Neuron. 2011 Mar 10;69(5):1038
Abstract
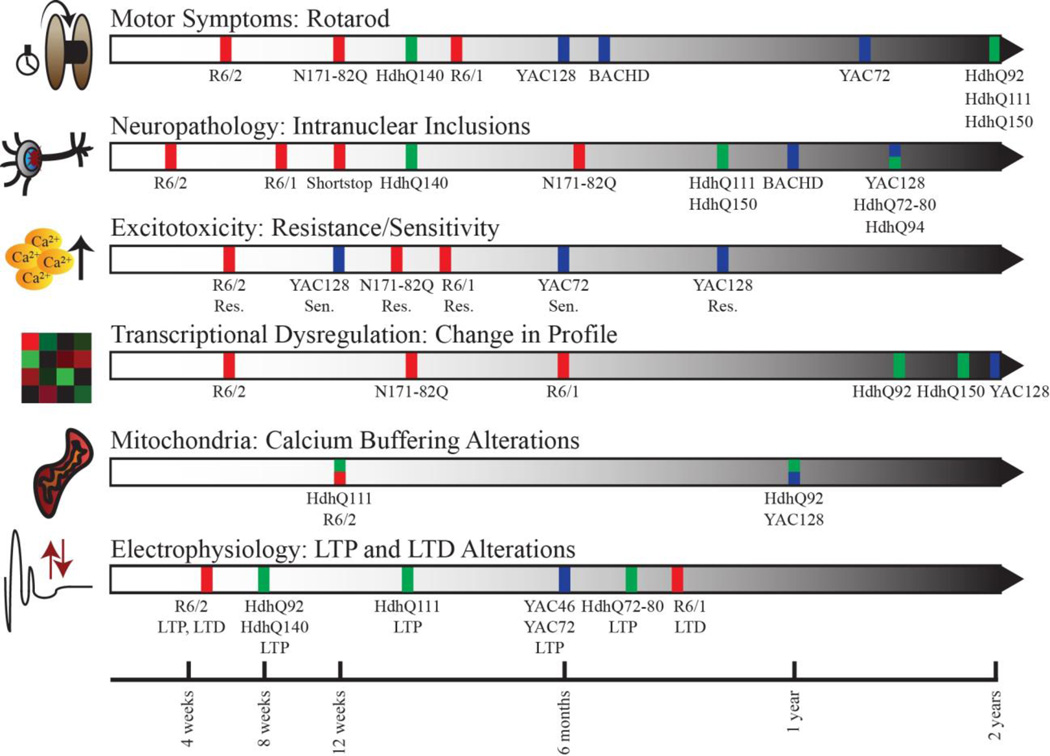
Mouse models for Huntington's Disease (HD) and HD patients demonstrate motor and behavioral dysfunctions, such as progressive loss of coordination and memory, and share similar transcriptional profiles and striatal neuron atrophy. Clear differences between the mouse and human diseases include almost complete striatal degeneration and rarity of intranuclear inclusions in HD, and the fact that mice expressing full-length mutant huntingtin do not demonstrate a shortened life span characteristic of HD. While no clinical interventions tested in mouse models to date have delayed disease progression, the mouse models provide an invaluable tool for both investigating the underlying pathogenic processes and developing new effective therapies. Inherent differences between humans and mice must be considered in the search for efficacious treatments for HD, but the striking similarities between human HD and mouse models support the view that these models are a biologically relevant system to support the identification and testing of potential clinical therapies.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, et al. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiology of Disease. 2001;8:479–491. - PubMed
-
- Bates G, Harper P, Jones L. Huntington’s Disease. 3rd edn. USA: Oxford University Press; 2002.
-
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–171. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
