

Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-γ
- PMID: 21317313
- PMCID: PMC3266063
- DOI: 10.1164/rccm.201009-1409OC
Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-γ
Abstract
Rationale: Diabetic patients have a lower incidence of acute respiratory distress syndrome (ARDS), and those who develop ARDS are less likely to die. The mechanisms that underlie this protection are unknown.
Objectives: To determine whether leptin resistance, a feature of diabetes, prevents fibroproliferation after lung injury.
Methods: We examined lung injury and fibroproliferation after the intratracheal instillation of bleomycin in wild-type and leptin-resistant (db/db) diabetic mice. We examined the effect of leptin on transforming growth factor (TGF)-β(1)-mediated transcription in primary normal human lung fibroblasts. Bronchoalveolar lavage fluid (BAL) samples from patients with ARDS and ventilated control subjects were obtained for measurement of leptin and active TGF-β(1) levels.
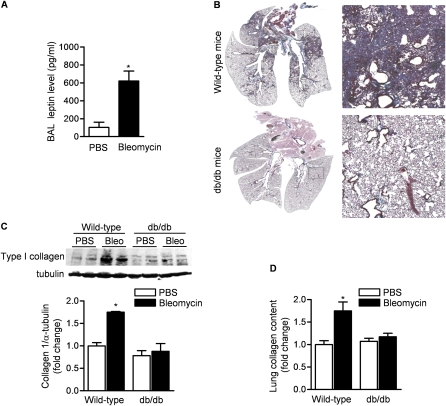
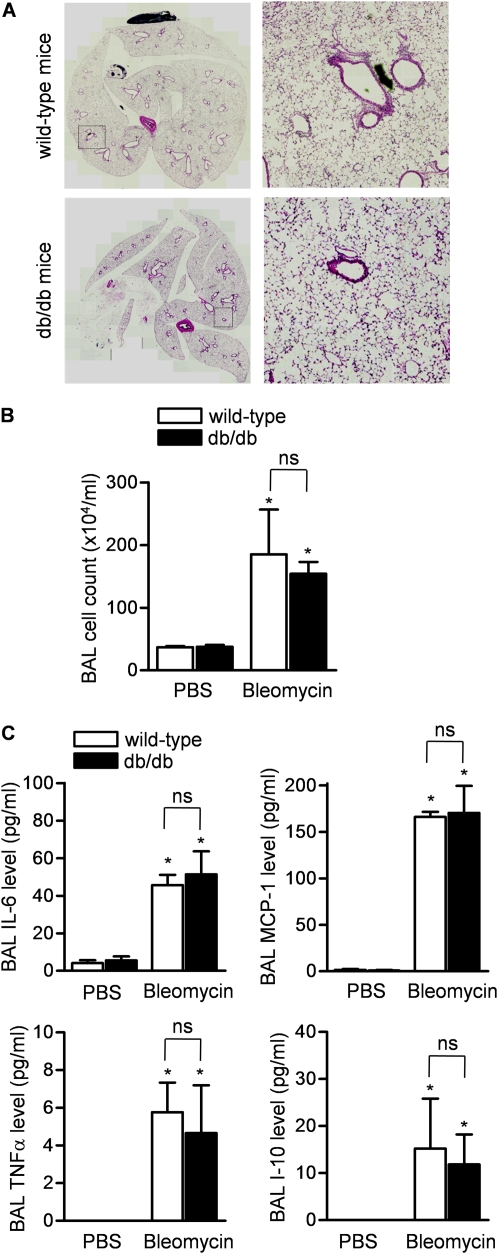
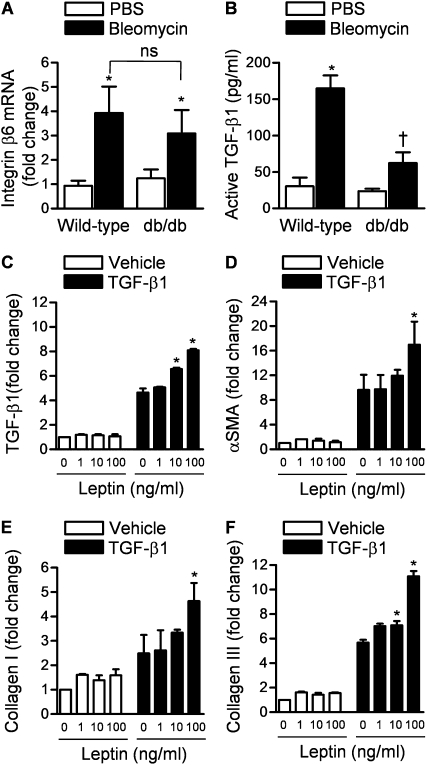
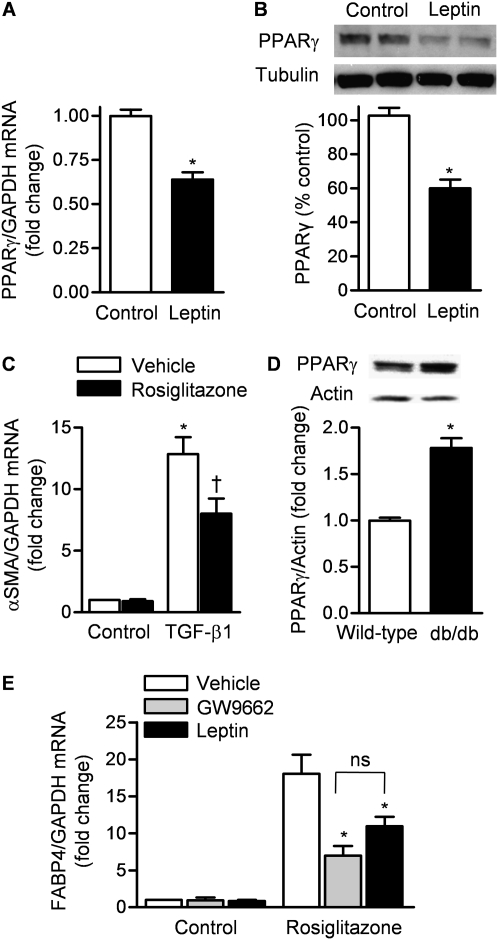
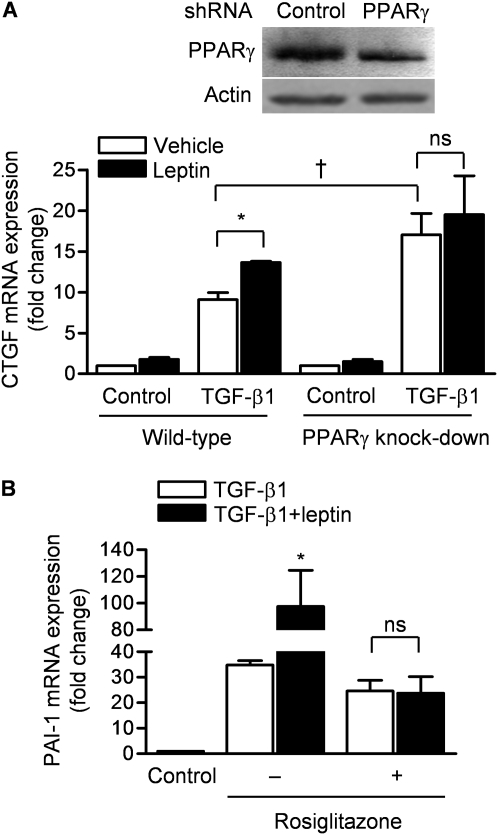
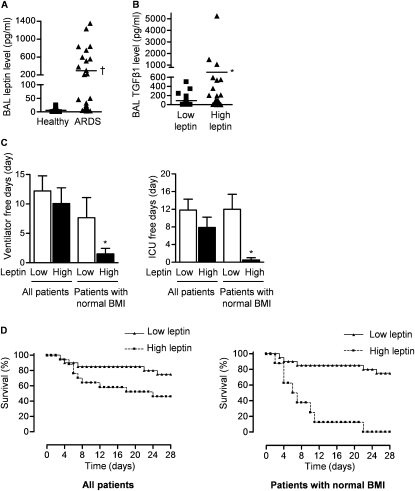
Measurements and main results: Diabetic mice (db/db) were resistant to lung fibrosis. The db/db mice had higher levels of peroxisome proliferator-activated receptor-γ (PPARγ), an inhibitor of the transcriptional response to TGF-β(1), a cytokine critical in the pathogenesis of fibroproliferative ARDS. In normal human lung fibroblasts, leptin augmented the transcription of profibrotic genes in response to TGF-β(1) through a mechanism that required PPARγ. In patients with ARDS, BAL leptin levels were elevated and correlated with TGF-β(1) levels. Overall, there was no significant relationship between BAL leptin levels and clinical outcomes; however, in nonobese patients, higher BAL leptin levels were associated with fewer intensive care unit- and ventilator-free days and higher mortality.
Conclusions: Leptin signaling is required for bleomycin-induced lung fibrosis. Leptin augments TGF-β(1) signaling in lung fibroblasts by inhibiting PPARγ. These findings provide a mechanism for the observed protection against ARDS observed in diabetic patients.
Figures
References
-
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353:1685–1693. - PubMed
-
- Moss M, Guidot DM, Steinberg KP, Duhon GF, Treece P, Wolken R, Hudson LD, Parsons PE. Diabetic patients have a decreased incidence of acute respiratory distress syndrome. Crit Care Med 2000;28:2187–2192. - PubMed
-
- Frank JA, Nuckton TJ, Matthay MA. Diabetes mellitus: a negative predictor for the development of acute respiratory distress syndrome from septic shock. Crit Care Med 2000;28:2645–2646. - PubMed
-
- Gajic O, Dabbagh O, Park PK, Adesanya A, Chang SY, Hou P, Anderson H III, Hoth JJ, Mikkelsen ME, Gentile NT, et al.; on behalf of the U.S. Critical Illness and Injury Trials Group: Lung Injury Prevention Study Investigators. Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med 2010;183:462–470. - PMC - PubMed
-
- Gong MN, Thompson BT, Williams P, Pothier L, Boyce PD, Christiani DC. Clinical predictors of and mortality in acute respiratory distress syndrome: potential role of red cell transfusion. Crit Care Med 2005;33:1191–1198. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
