

Factors determining DNA double-strand break repair pathway choice in G2 phase
- PMID: 21317870
- PMCID: PMC3061033
- DOI: 10.1038/emboj.2011.27
Factors determining DNA double-strand break repair pathway choice in G2 phase
Abstract
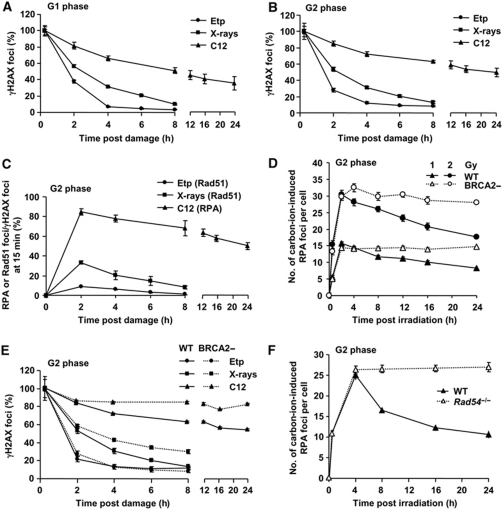
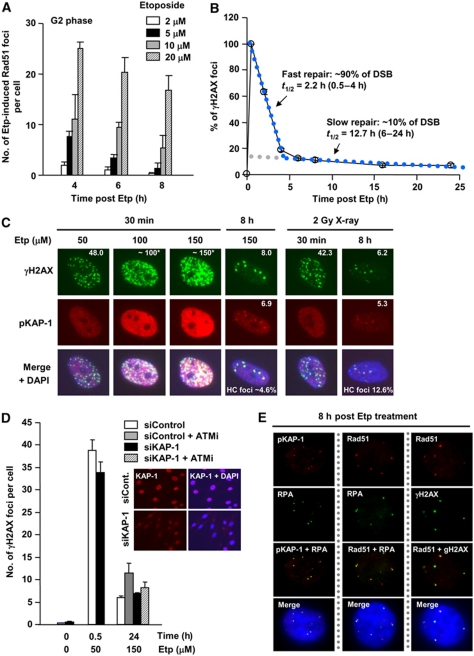
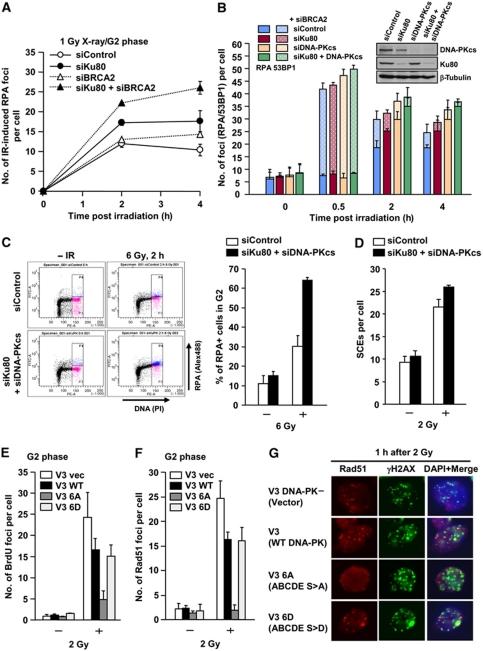
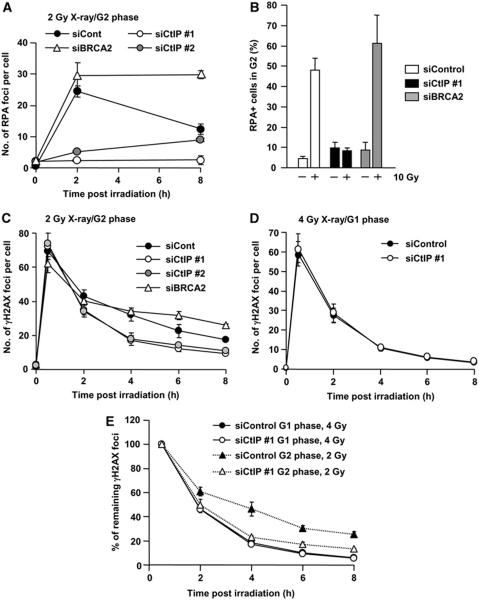
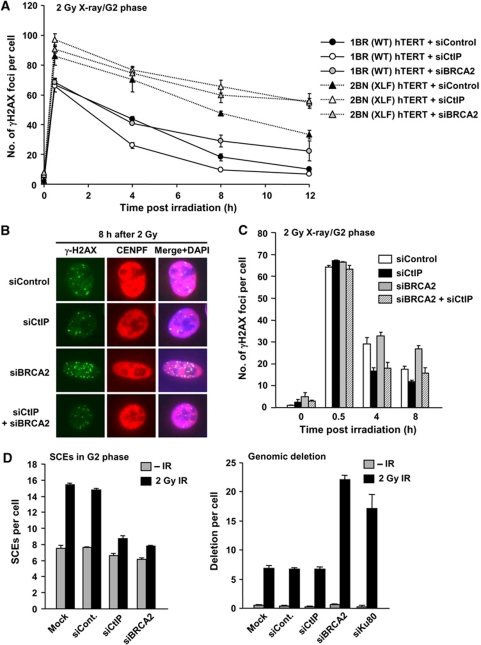
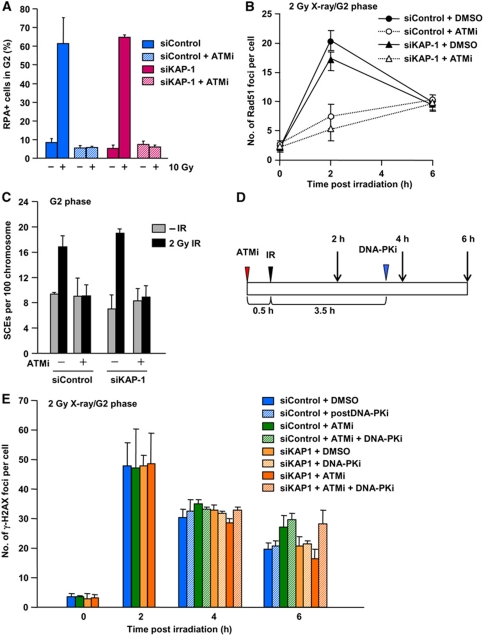
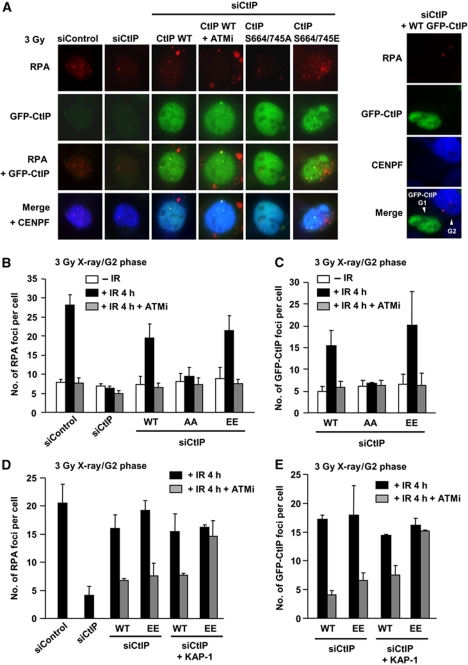
DNA non-homologous end joining (NHEJ) and homologous recombination (HR) function to repair DNA double-strand breaks (DSBs) in G2 phase with HR preferentially repairing heterochromatin-associated DSBs (HC-DSBs). Here, we examine the regulation of repair pathway usage at two-ended DSBs in G2. We identify the speed of DSB repair as a major component influencing repair pathway usage showing that DNA damage and chromatin complexity are factors influencing DSB repair rate and pathway choice. Loss of NHEJ proteins also slows DSB repair allowing increased resection. However, expression of an autophosphorylation-defective DNA-PKcs mutant, which binds DSBs but precludes the completion of NHEJ, dramatically reduces DSB end resection at all DSBs. In contrast, loss of HR does not impair repair by NHEJ although CtIP-dependent end resection precludes NHEJ usage. We propose that NHEJ initially attempts to repair DSBs and, if rapid rejoining does not ensue, then resection occurs promoting repair by HR. Finally, we identify novel roles for ATM in regulating DSB end resection; an indirect role in promoting KAP-1-dependent chromatin relaxation and a direct role in phosphorylating and activating CtIP.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Chan DW, Lees-Miller SP (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem 271: 8936–8941 - PubMed
-
- Drouet J, Delteil C, Lefrancois J, Concannon P, Salles B, Calsou P (2005) DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem 280: 7060–7069 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
