

Glycogen storage disease type Ia in canines: a model for human metabolic and genetic liver disease
- PMID: 21318173
- PMCID: PMC3027000
- DOI: 10.1155/2011/646257
Glycogen storage disease type Ia in canines: a model for human metabolic and genetic liver disease
Abstract
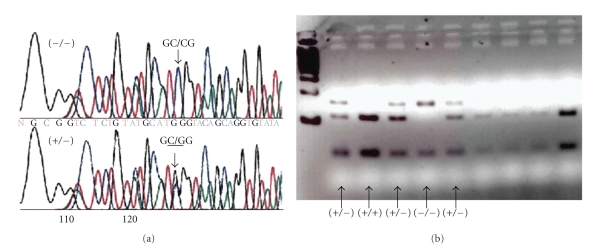
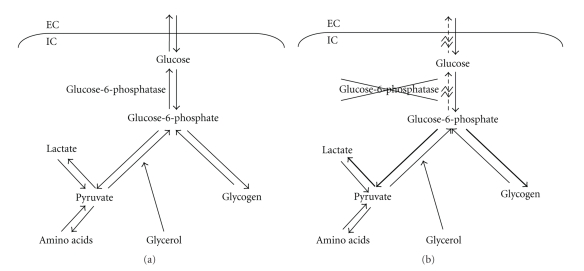
A canine model of Glycogen storage disease type Ia (GSDIa) is described. Affected dogs are homozygous for a previously described M121I mutation resulting in a deficiency of glucose-6-phosphatase-α. Metabolic, clinicopathologic, pathologic, and clinical manifestations of GSDIa observed in this model are described and compared to those observed in humans. The canine model shows more complete recapitulation of the clinical manifestations seen in humans including "lactic acidosis", larger size, and longer lifespan compared to other animal models. Use of this model in preclinical trials of gene therapy is described and briefly compared to the murine model. Although the canine model offers a number of advantages for evaluating potential therapies for GSDIa, there are also some significant challenges involved in its use. Despite these challenges, the canine model of GSDIa should continue to provide valuable information about the potential for generating curative therapies for GSDIa as well as other genetic hepatic diseases.
Figures
References
-
- Chen YT, Burchell A. Glycogen storage diseases. In: Schriver C, Beaudet A, editors. The Metabolic and Molecular Basis of Inherited Disease. New York, NY, USA: McGraw-Hill; 1995. pp. 905–934.
-
- Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Current Molecular Medicine. 2002;2(2):121–143. - PubMed
-
- Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Reviews in Endocrine and Metabolic Disorders. 2003;4(1):95–102. - PubMed
-
- Smit GPA, Berger R, Potasnick R. The dietary treatment of children with type I glycogen storage disease with slow release carbohydrate. Pediatric Research. 1984;18(9):879–881. - PubMed
-
- Smit GPA, Ververs MT, Belderok B, Van Rijn M, Berger R, Fernandes J. Complex carbohydrates in the dietary management of patients with glycogenosis caused by glucose-6-phosphate deficiency. American Journal of Clinical Nutrition. 1988;48(1):95–97. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
