

doi: 10.1093/bioinformatics/btr076.
Epub 2011 Feb 13.
Improving SNP discovery by base alignment quality
Affiliations
- PMID: 21320865
- PMCID: PMC3072548
- DOI: 10.1093/bioinformatics/btr076
Item in Clipboard
Improving SNP discovery by base alignment quality
Bioinformatics.
.
Abstract
I propose a new application of profile Hidden Markov Models in the area of SNP discovery from resequencing data, to greatly reduce false SNP calls caused by misalignments around insertions and deletions (indels). The central concept is per-Base Alignment Quality, which accurately measures the probability of a read base being wrongly aligned. The effectiveness of BAQ has been positively confirmed on large datasets by the 1000 Genomes Project analysis subgroup.
Availability: http://samtools.sourceforge.net
Contact: hengli@broadinstitute.org.
Figures
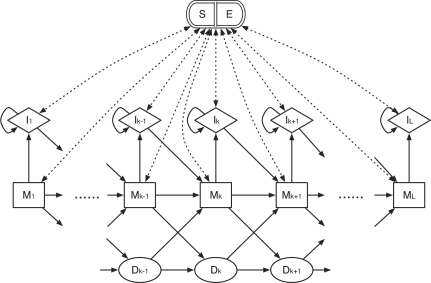
The topology of the profile HMM for BAQ computation. It consists of five types of states: alignment matches (M), insertions to the reference (I), deletions (D), alignment start (S) and alignment end (E). The S state points to every M and I state while every M and I points to E. States S and E are plotted together to avoid excessive dotted lines in the figure.
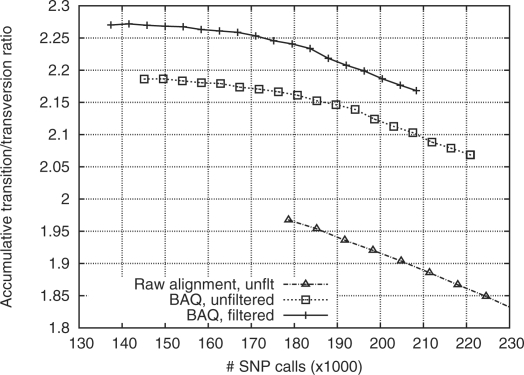
Transition–transversion ratio (ts/tv) as a function of the number of SNP calls. SNPs are sorted by the posterior probability of the site being a SNP (SNP probability). Given a threshold on the SNP probability, the number of SNPs of higher probability and their ts/tv are plotted. For the solid line, filters in use are as follows: (i) total depth below 500; and (ii) root mean square mapping quality above 10; (iii) P-value of reference and non-reference bases being evenly distributed on both strands is above 10−4 (by exact test).
References
-
- Durbin R., et al. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
