

PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia
- PMID: 21321125
- PMCID: PMC3075643
- DOI: 10.1074/jbc.M110.204024
PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia
Abstract
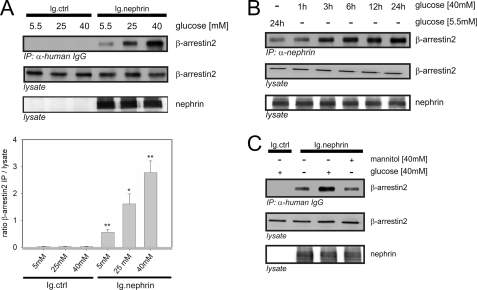
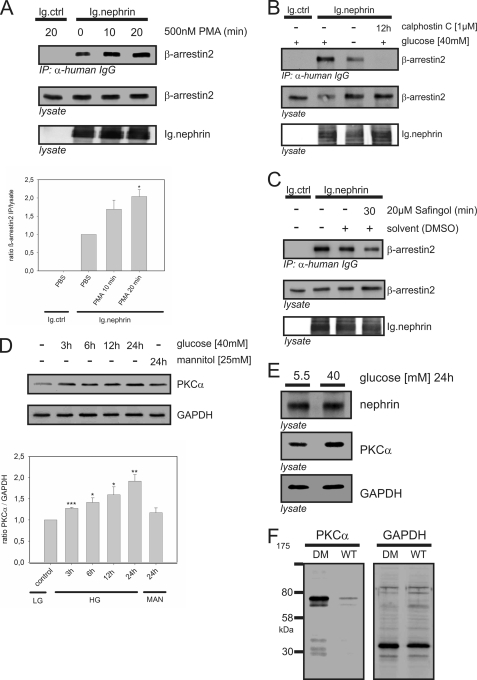
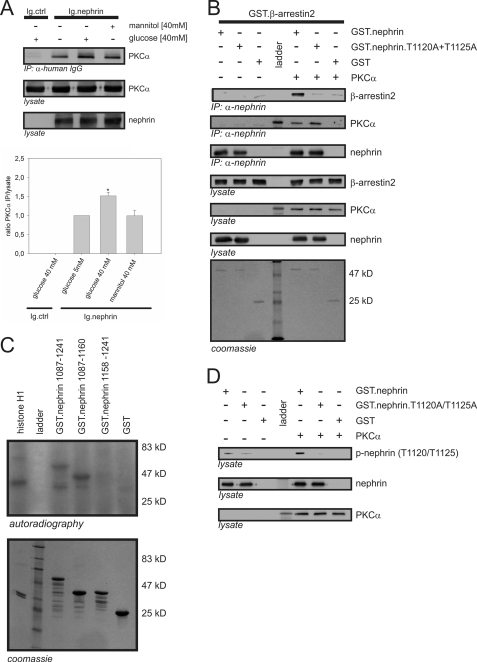
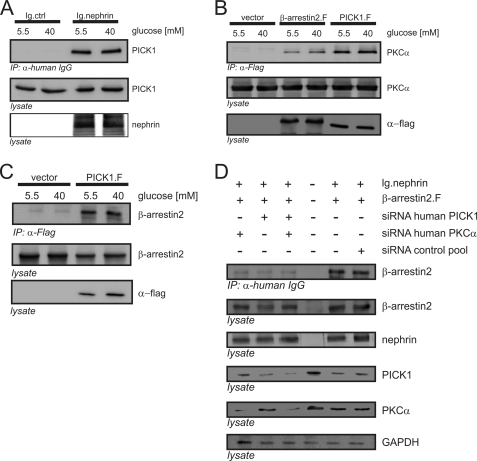
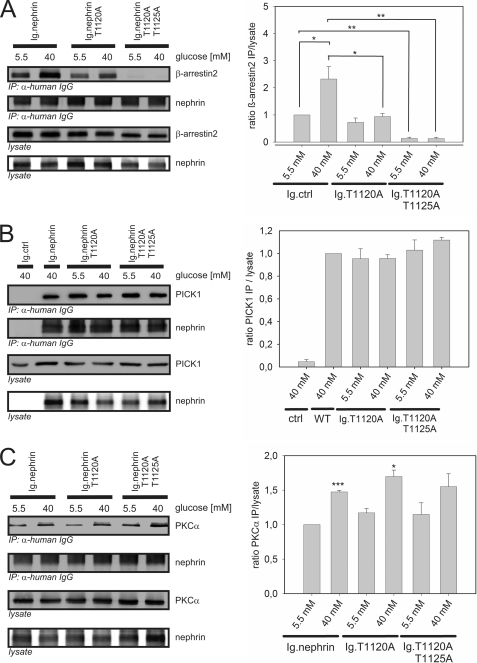
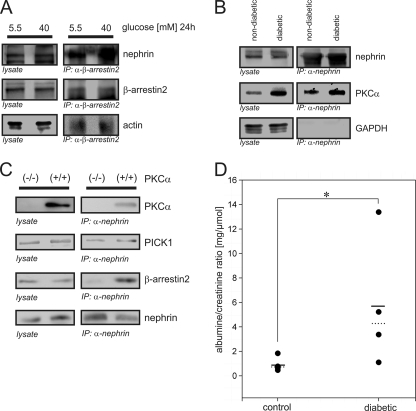
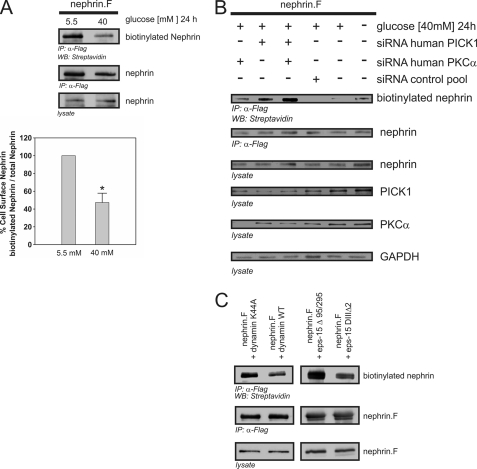
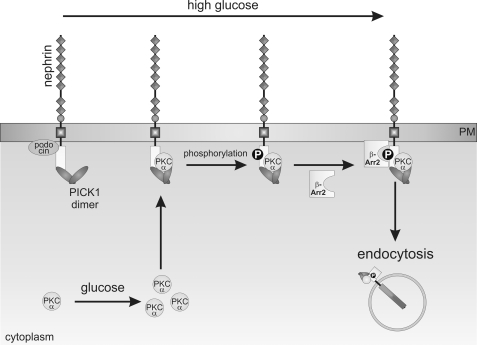
Nephrin, the key molecule of the glomerular slit diaphragm, is expressed on the surface of podocytes and is critical in preventing albuminuria. In diabetes, hyperglycemia leads to the loss of surface expression of nephrin and causes albuminuria. Here, we report a mechanism that can explain this phenomenon: hyperglycemia directly enhances the rate of nephrin endocytosis via regulation of the β-arrestin2-nephrin interaction by PKCα. We identified PKCα and protein interacting with c kinase-1 (PICK1) as nephrin-binding proteins. Hyperglycemia induced up-regulation of PKCα and led to the formation of a complex of nephrin, PKCα, PICK1, and β-arrestin2 in vitro and in vivo. Binding of β-arrestin2 to the nephrin intracellular domain depended on phosphorylation of nephrin threonine residues 1120 and 1125 by PKCα. Further, cellular knockdown of PKCα and/or PICK1 attenuated the nephrin-β-arrestin2 interaction and abrogated the amplifying effect of high blood glucose on nephrin endocytosis. In C57BL/6 mice, hyperglycemia over 24 h caused a significant increase in urinary albumin excretion, supporting the concept of the rapid impact of hyperglycemia on glomerular permselectivity. In summary, we have provided a molecular model of hyperglycemia-induced nephrin endocytosis and subsequent proteinuria and highlighted PKCα and PICK1 as promising therapeutic targets for diabetic nephropathy.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
