

DeltAMT: a statistical algorithm for fast detection of protein modifications from LC-MS/MS data
- PMID: 21321130
- PMCID: PMC3098578
- DOI: 10.1074/mcp.M110.000455
DeltAMT: a statistical algorithm for fast detection of protein modifications from LC-MS/MS data
Abstract
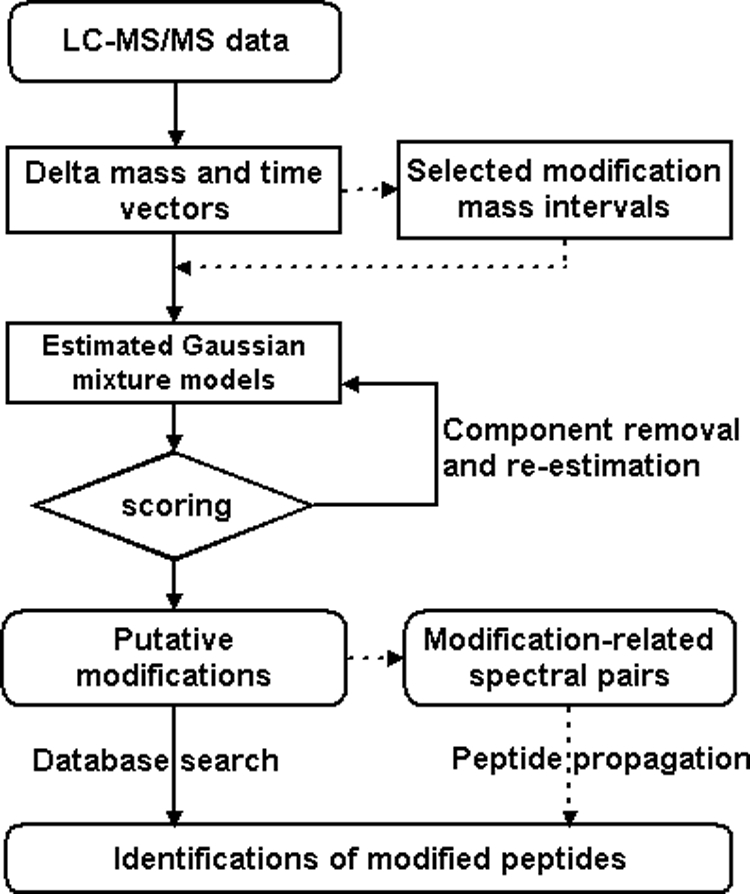
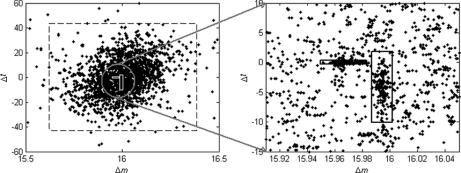
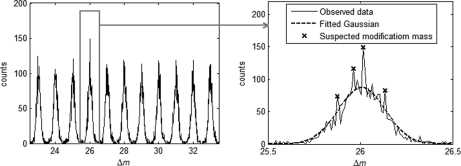
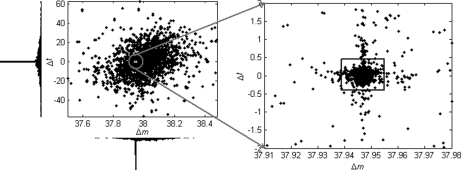
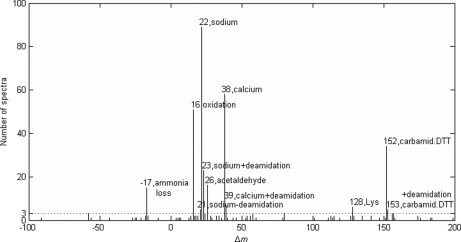
Identification of proteins and their modifications via liquid chromatography-tandem mass spectrometry is an important task for the field of proteomics. However, because of the complexity of tandem mass spectra, the majority of the spectra cannot be identified. The presence of unanticipated protein modifications is among the major reasons for the low spectral identification rate. The conventional database search approach to protein identification has inherent difficulties in comprehensive detection of protein modifications. In recent years, increasing efforts have been devoted to developing unrestrictive approaches to modification identification, but they often suffer from their lack of speed. This paper presents a statistical algorithm named DeltAMT (Delta Accurate Mass and Time) for fast detection of abundant protein modifications from tandem mass spectra with high-accuracy precursor masses. The algorithm is based on the fact that the modified and unmodified versions of a peptide are usually present simultaneously in a sample and their spectra are correlated with each other in precursor masses and retention times. By representing each pair of spectra as a delta mass and time vector, bivariate Gaussian mixture models are used to detect modification-related spectral pairs. Unlike previous approaches to unrestrictive modification identification that mainly rely upon the fragment information and the mass dimension in liquid chromatography-tandem mass spectrometry, the proposed algorithm makes the most of precursor information. Thus, it is highly efficient while being accurate and sensitive. On two published data sets, the algorithm effectively detected various modifications and other interesting events, yielding deep insights into the data. Based on these discoveries, the spectral identification rates were significantly increased and many modified peptides were identified.
Figures
References
-
- Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 - PubMed
-
- Mann M., Jensen O. N. (2003) Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 - PubMed
-
- Witze E. S., Old W. M., Resing K. A., Ahn N. G. (2007) Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806 - PubMed
-
- Eng J. K., McCormack A. L., Yates J. R., III (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass. Spectrom. 5, 976–989 - PubMed
-
- Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
