

doi: 10.1186/gb-2011-12-2-r15.
Epub 2011 Feb 16.
A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets
Affiliations
- PMID: 21324173
- PMCID: PMC3188797
- DOI: 10.1186/gb-2011-12-2-r15
Item in Clipboard
A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets
Genome Biol.
2011.
Abstract
We develop a statistical framework to study the relationship between chromatin features and gene expression. This can be used to predict gene expression of protein coding genes, as well as microRNAs. We demonstrate the prediction in a variety of contexts, focusing particularly on the modENCODE worm datasets. Moreover, our framework reveals the positional contribution around genes (upstream or downstream) of distinct chromatin features to the overall prediction of expression levels.
Figures
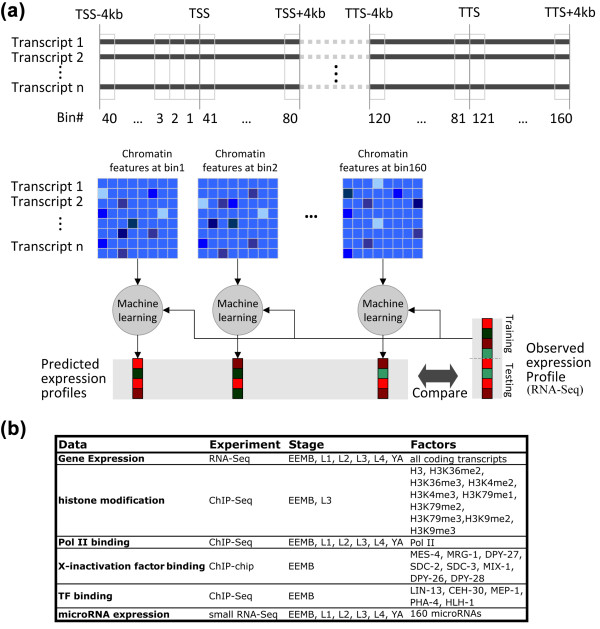
Schematic diagram of our data binning and supervised analysis. (a) DNA regions around the transcription start site (TSS) and transcription terminal site (TTS) of each transcript were separated into 160 bins of 100 bp in size. Average signal of each chromatin feature was calculated for all transcripts, resulting in a predictor matrix for each bin. These predictor matrices were used to predict expression of transcripts by support vector machine (SVM) or support vector regression (SVR) models. The genome-wide data for chromatin features and gene expression were generated by the modENCODE project using ChIP-chip/ChIP-seq and RNA-seq experiments, respectively. (b) A summary of datasets used in our analysis. L, larval; TF, transcription factor; YA, young adult.
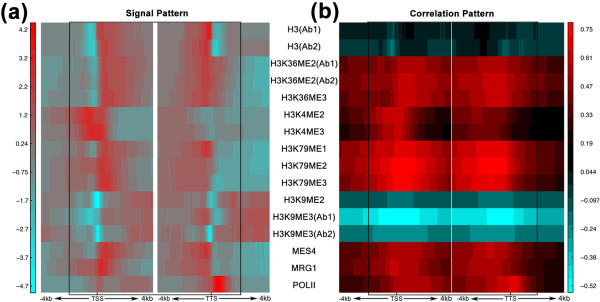
Chromatin feature patterns. (a,b) Signal pattern (a) and correlation pattern (b) of each chromatin feature in the 160 bins around the TSS and TTS (from 4 kb upstream to 4 kb downstream) of worm transcripts at the EEMB stage. In (a), the signal of each chromatin feature for each bin is averaged across all transcripts. In (b), the Spearman correlation coefficient of each chromatin feature with gene expression levels was calculated for each bin. Ab1 and Ab2 represent experimental results using different antibodies for a chromatin feature. DNA region from 2 kb upstream of the TSS to 2 kb downstream of the TTS is shown in the rectangle.
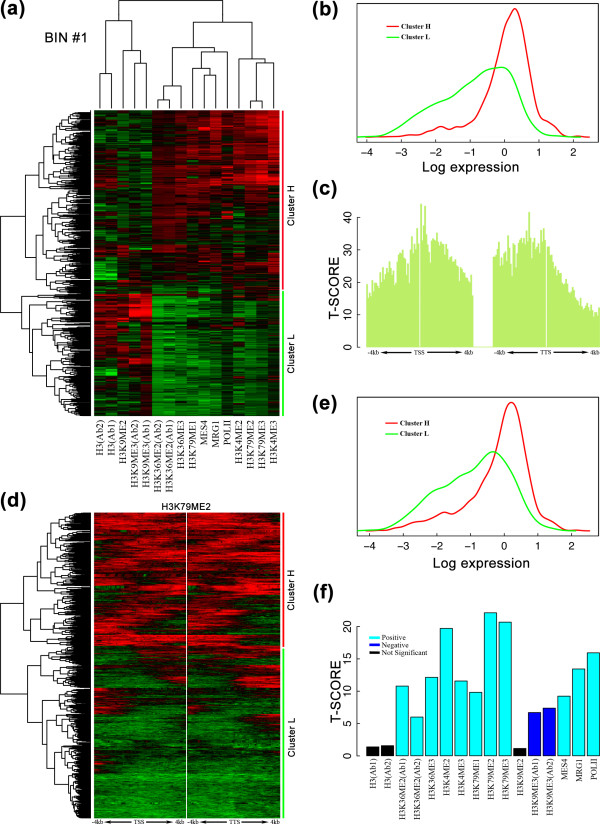
Hierarchical clustering using either chromatin feature profiles (a-c) or bin profiles (d-f) discriminates highly and lowly expressed genes. (a) Hierarchical clustering of 16 chromatin features in bin 1 (0 to 100 nucleotides upstream of a TSS). The resulting tree is split at the top branch, which divides genes into two clusters, cluster H and cluster L, as labeled. (b) Distributions of expression levels of genes in cluster H (red) and cluster L (green). Expression levels are significantly different between the two clusters according to t-test (P = 3E-202). Expression levels were measured by RNA-seq (see Materials and methods). (c) T-scores for the differential expression of the top two gene clusters based on hierarchical clustering of chromatin features in each of the 160 bins. For each bin, hierarchical clustering was performed to separate genes into two clusters. Expression levels between the two clusters were compared and a t-score calculated to measure the capability of the bin to discriminate between genes with high and low expression levels. (d) Hierarchical clustering of the genes based on the signal profiles of H3K79me2 across the 160 bins. The resulting tree is also split at the top branch, leading to two gene clusters. (e) Distributions of expression levels of genes in the two clusters in (d). The expression levels are significantly different according to t-test (P = 4E-93). (f) T-scores for the differential expression of the two gene clusters based on hierarchical clustering of bin profiles for each individual chromatin feature. Cyan and blue colors indicate a significant positive and negative correlation between a chromatin feature and gene expression levels, respectively. Black color indicates that a chromatin feature could not significantly discriminate between genes with high and low expression levels. To visualize the clustering, 2,000 randomly selected genes are shown. The data for gene expression levels and chromatin features are from the EEMB stage.
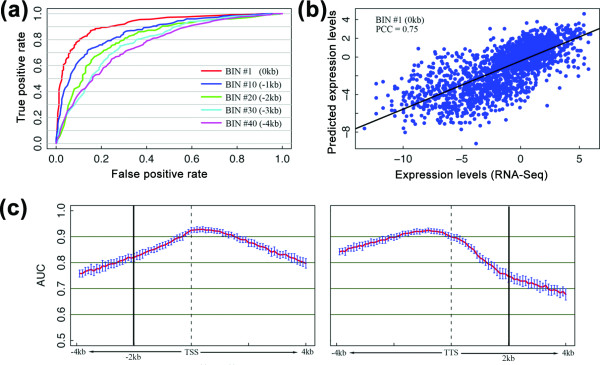
Prediction power of the supervised models. (a) ROC curves for five different bins based on the results of the SVM classification models. (b) Predicted versus experimentally measured expression levels. The SVR regression model was applied to bin 1 for predicting gene expression levels. (PCC, Pearson correlation coefficient). (c) The prediction accuracy of SVM classification models for all the 160 bins. For each bin, we constructed an SVM classification model and summarized its accuracy using the AUC score. The AUC scores were calculated based on cross-validation repeated 100 times for each bin. The red curve shows the average AUC scores (mean of 100 repeats) of the bins and the blue bars indicate their standard deviations. The positions of the TSS and TTS are marked by dotted lines.
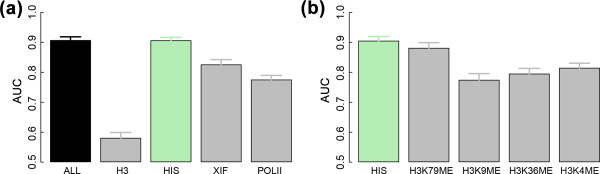
Prediction power of the SVM models using the signals from different subsets of chromatin features in the 100 nucleotides around the TSS (bin 1). The results are based on cross-validation with 100 trials. (a) ALL, all 21 chromatin features; H3, the two H3 features; HIS, the 11 chromatin modification features; XIF, the seven binding profile features for X-inactivation factors; POLII, the binding profile feature for RNA polymerase II. (b) HIS, the 11 chromatin modification features; H3K79ME, H3K79me1, H3K79me2 and H3K79me3; H3K9ME, H3K9me2, H3K9me3(Ab1) and H3K9me3(Ab2); H3K36ME, H3K36me2(Ab1), H3K36me2(Ab2) and H3K36me3; H3K4ME, H3K4me3 and H3K4me3.
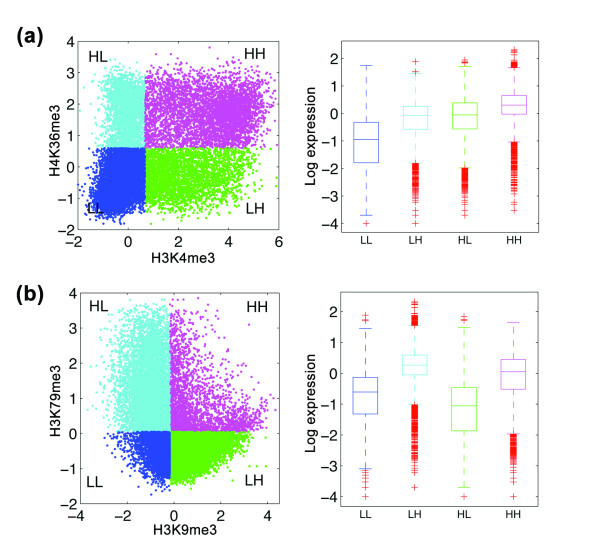
Co-regulation of transcription by pairs of histone modifications. (a) Categorization of genes into four groups based on signals of H3K4me3 and H3K36me3: HH (magenta), HL (green), LH (cyan) and LL (blue). The signals of histone marks H3K36me3 and H3K4me3 exhibit a bimodal feature. Signals are thus classified into H and L by a Gaussian mixture model. The distributions of expression levels of the four gene groups are shown on the right. (b) Same as (a), based on signals of H3K9me3 and H3K79me3. Same as above, the signal of H3K79me3 is again classified by a Gaussian mixture model. The signals of H3K9me3 do not display a bimodal feature; signals are classified into H and L based on whether the value is higher than or lower than the median.
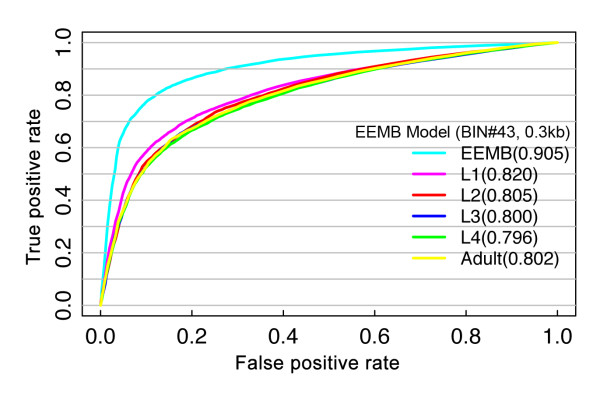
Developmental stage specificity of the chromatin model. The EEMB model was constructed using the chromatin features and gene expression data both at the EEMB stage. The model was then used to predict gene expression levels at the EEMB stage and five other developmental stages: L1, L2, L3, L4 and adult. ROC curves are plotted based on the results of 100 trials of cross-validation. For each trial, the dataset was randomly separated into two halves: one half as training data and the other as testing data to estimate the accuracy of the model. The values in parentheses are AUC scores.
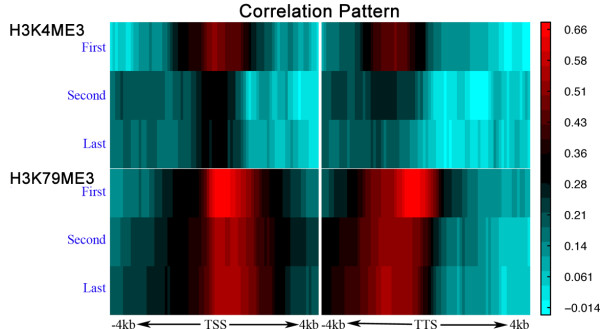
Correlation patterns of H3K4me3 and H3K79me3 in the 160 bins around the TSS and TTS (from 4 kb upstream to 4 kb downstream) with the expression levels of the first, second and last genes of 881 C. elegans operons.
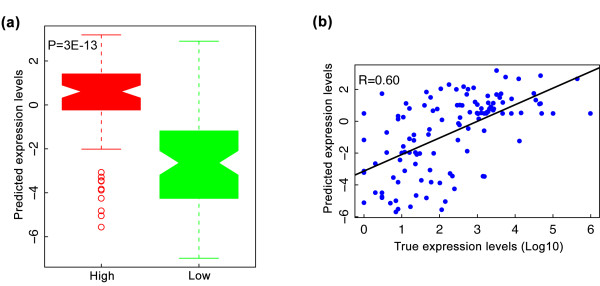
Prediction of expression levels of microRNAs at the EEMB stage. (a) Predicted expression levels of the experimentally measured highly and lowly expressed microRNAs based on small RNA-seq results. Expression levels of microRNAs at the EEMB stage were predicted using an SVR regression model trained on data for protein-coding genes at the same stage. (b) Predicted versus experimentally measured expression levels of microRNAs at the EEMB stage. R is the Pearson correlation coefficient.
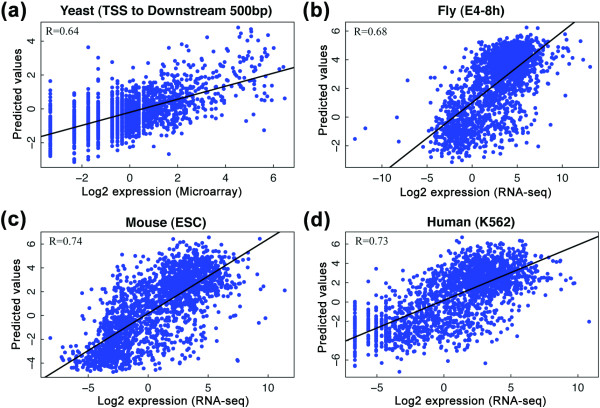
Prediction accuracy of the chromatin model in four other species. (a-d) Expression levels of genes are predicted using the SVR method. In yeast, average signals of chromatin features from the TSS to 500 bp upstream were used as predictors (a); in the other species, signals of chromatin features within the bin at the TSS (bin 1) were used as predictors (b-d). E4-8 h: embryonic stage at 4 to 8 h; ESC, embryonic stem cell.
References
-
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H. et al.Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
