

Genomewide characterization of non-polyadenylated RNAs
- PMID: 21324177
- PMCID: PMC3188798
- DOI: 10.1186/gb-2011-12-2-r16
Genomewide characterization of non-polyadenylated RNAs
Abstract
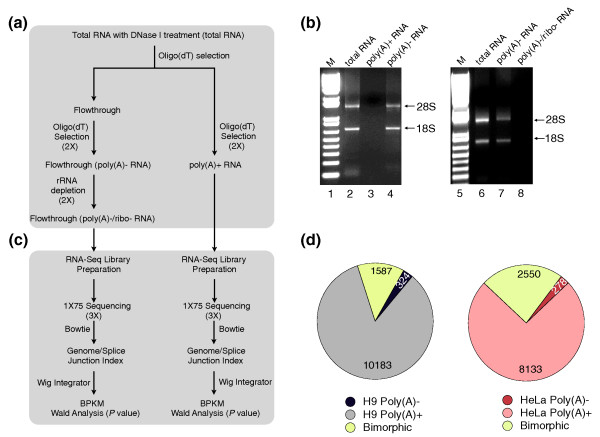
Background: RNAs can be physically classified into poly(A)+ or poly(A)- transcripts according to the presence or absence of a poly(A) tail at their 3' ends. Current deep sequencing approaches largely depend on the enrichment of transcripts with a poly(A) tail, and therefore offer little insight into the nature and expression of transcripts that lack poly(A) tails.
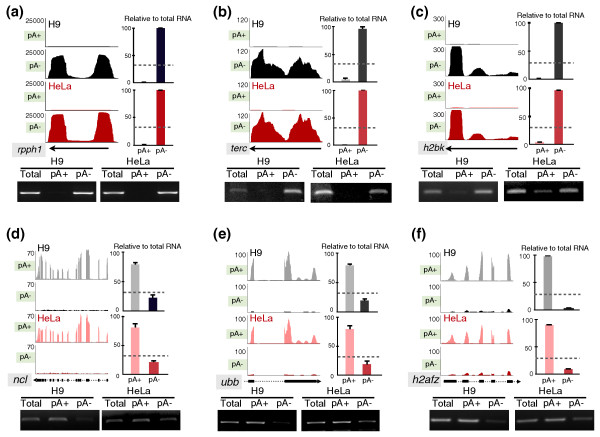
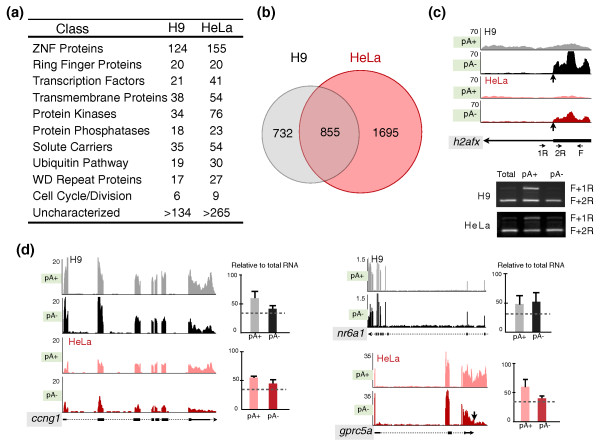
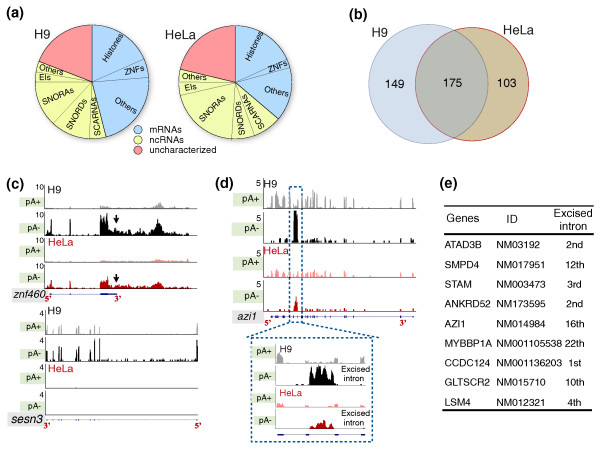
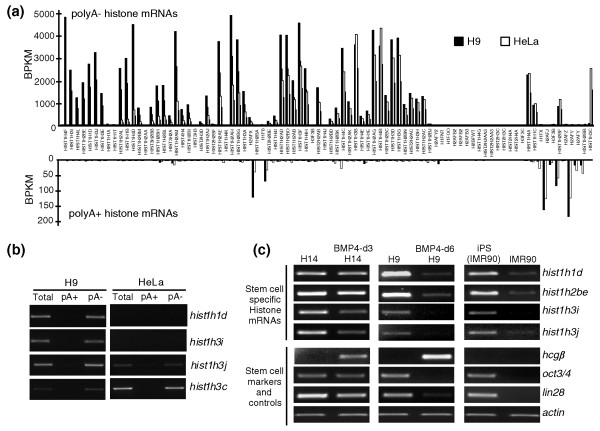
Results: We have used deep sequencing to explore the repertoire of both poly(A)+ and poly(A)- RNAs from HeLa cells and H9 human embryonic stem cells (hESCs). Using stringent criteria, we found that while the majority of transcripts are poly(A)+, a significant portion of transcripts are either poly(A)- or bimorphic, being found in both the poly(A)+ and poly(A)- populations. Further analyses revealed that many mRNAs may not contain classical long poly(A) tails and such messages are overrepresented in specific functional categories. In addition, we surprisingly found that a few excised introns accumulate in cells and thus constitute a new class of non-polyadenylated long non-coding RNAs. Finally, we have identified a specific subset of poly(A)- histone mRNAs, including two histone H1 variants, that are expressed in undifferentiated hESCs and are rapidly diminished upon differentiation; further, these same histone genes are induced upon reprogramming of fibroblasts to induced pluripotent stem cells.
Conclusions: We offer a rich source of data that allows a deeper exploration of the poly(A)- landscape of the eukaryotic transcriptome. The approach we present here also applies to the analysis of the poly(A)- transcriptomes of other organisms.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
