

Maximum-likelihood estimation of recent shared ancestry (ERSA)
- PMID: 21324875
- PMCID: PMC3083094
- DOI: 10.1101/gr.115972.110
Maximum-likelihood estimation of recent shared ancestry (ERSA)
Abstract
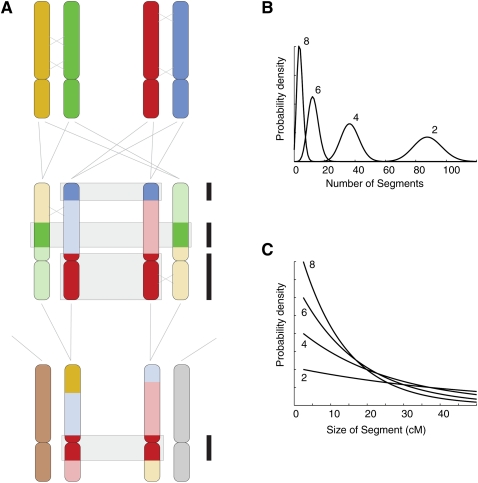
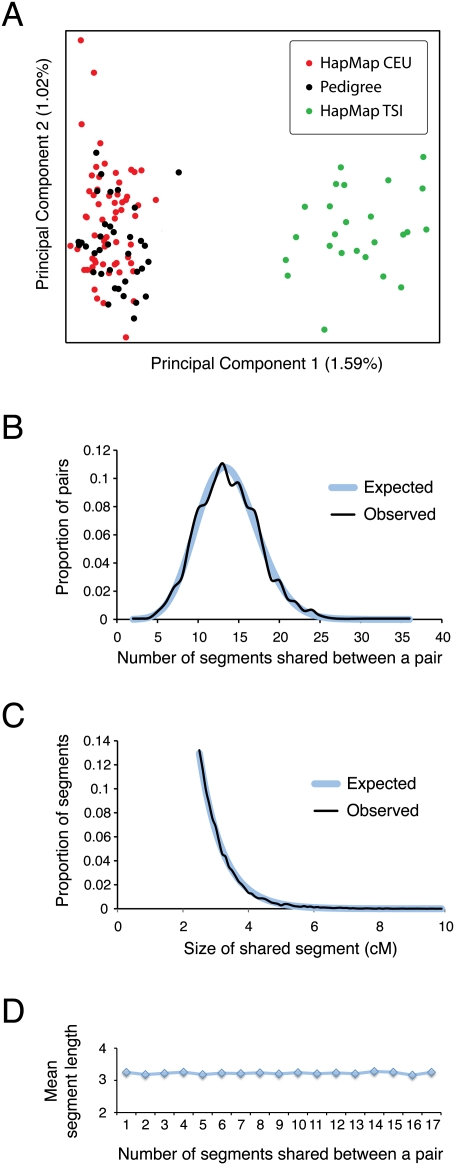
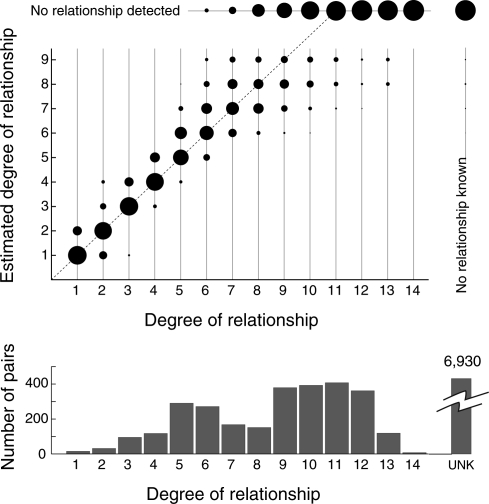
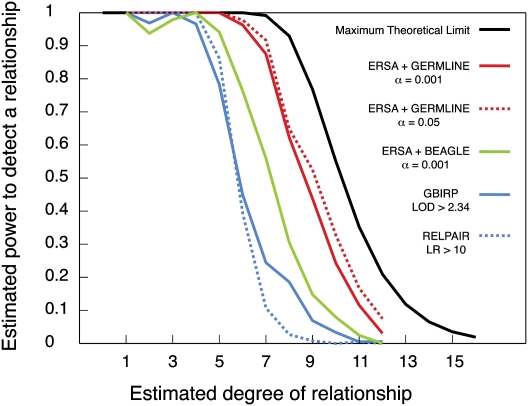
Accurate estimation of recent shared ancestry is important for genetics, evolution, medicine, conservation biology, and forensics. Established methods estimate kinship accurately for first-degree through third-degree relatives. We demonstrate that chromosomal segments shared by two individuals due to identity by descent (IBD) provide much additional information about shared ancestry. We developed a maximum-likelihood method for the estimation of recent shared ancestry (ERSA) from the number and lengths of IBD segments derived from high-density SNP or whole-genome sequence data. We used ERSA to estimate relationships from SNP genotypes in 169 individuals from three large, well-defined human pedigrees. ERSA is accurate to within one degree of relationship for 97% of first-degree through fifth-degree relatives and 80% of sixth-degree and seventh-degree relatives. We demonstrate that ERSA's statistical power approaches the maximum theoretical limit imposed by the fact that distant relatives frequently share no DNA through a common ancestor. ERSA greatly expands the range of relationships that can be estimated from genetic data and is implemented in a freely available software package.
Figures
References
-
- Alonso A, Martin P, Albarran C, Garcia P, Fernandez de Simon L, Jesus Iturralde M, Fernandez-Rodriguez A, Atienza I, Capilla J, Garcia-Hirschfeld J, et al. 2005. Challenges of DNA profiling in mass disaster investigations. Croat Med J 46: 540–548 - PubMed
-
- Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, Lullmann-Rauch R, Blanz J, Zhang KW, Stankovich J, et al. 2008. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 82: 673–684 - PMC - PubMed
-
- Bieber FR, Brenner CH, Lazer D 2006. Finding criminals through DNA of their relatives. Science 312: 1315–1316 - PubMed
-
- Biesecker LG, Bailey-Wilson JE, Ballantyne J, Baum H, Bieber FR, Brenner C, Budowle B, Butler JM, Carmody G, Conneally PM, et al. 2005. Epidemiology. DNA identifications after the 9/11 World Trade Center attack. Science 310: 1122–1123 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources