

Thioredoxin increases exocytosis by denitrosylating N-ethylmaleimide-sensitive factor
- PMID: 21324905
- PMCID: PMC3064172
- DOI: 10.1074/jbc.M110.201780
Thioredoxin increases exocytosis by denitrosylating N-ethylmaleimide-sensitive factor
Abstract
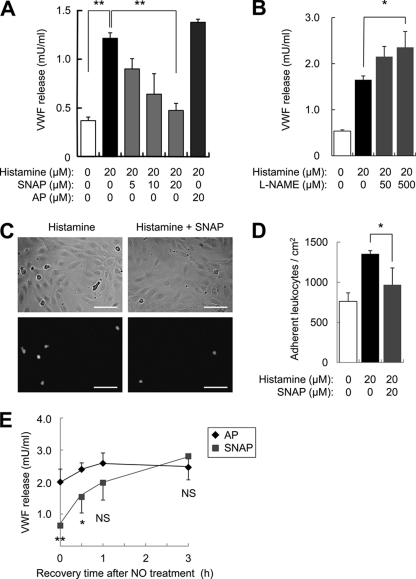
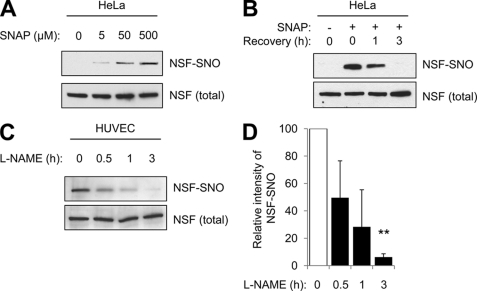
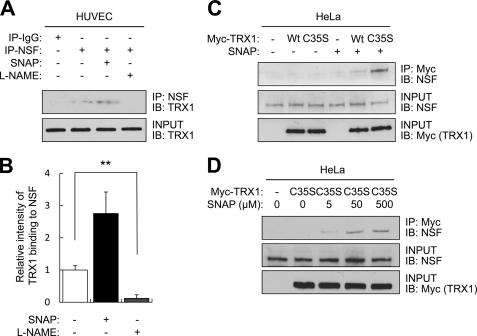
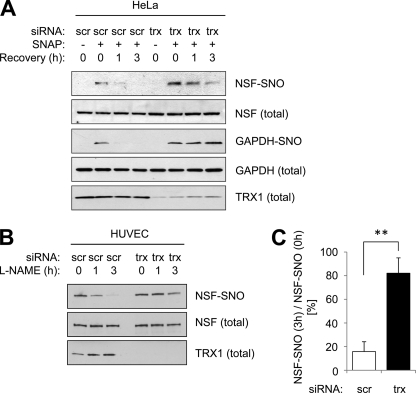
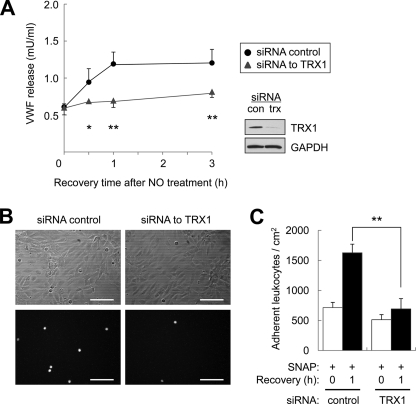
Exocytosis involves membrane fusion between granules and the plasma membrane. Nitric oxide (NO) inhibits exocytosis by chemically modifying N-ethylmaleimide-sensitive factor (NSF), a key component of the exocytic machinery. However, cells recover the ability to release messenger molecules within hours of exposure to NO through unknown mechanisms. We now identify thioredoxin (TRX1) as a denitrosylase that reverses NO inhibition of exocytosis. Endogenously synthesized NO increases S-nitrosylated NSF levels, but S-nitrosylated NSF levels decrease within 3 h after exposure to NO. We found that NO increases the interaction between TRX1 and NSF, and endogenous TRX1 removes NO from S-nitrosylated NSF. Knockdown of TRX1 increases the level of S-nitrosylated NSF, prolongs the inhibition of exocytosis, and suppresses leukocyte adhesion. Taken together, these data show that TRX1 promotes exocytosis by denitrosylating NSF. Our findings suggest that TRX1 might regulate exocytosis in a variety of physiological settings, such as vascular inflammation, thrombosis, and insulin release.
Figures
Similar articles
-
N-ethylmaleimide-sensitive factor: a redox sensor in exocytosis.Biol Chem. 2006 Oct-Nov;387(10-11):1377-83. doi: 10.1515/BC.2006.173. Biol Chem. 2006. PMID: 17081110 Review.
-
Exocytosis of endothelial cells is regulated by N-ethylmaleimide-sensitive factor.Methods Mol Biol. 2008;440:203-15. doi: 10.1007/978-1-59745-178-9_15. Methods Mol Biol. 2008. PMID: 18369947 Free PMC article.
-
Nitric oxide regulation of protein trafficking in the cardiovascular system.Cardiovasc Res. 2007 Jul 15;75(2):240-6. doi: 10.1016/j.cardiores.2007.03.024. Epub 2007 Apr 3. Cardiovasc Res. 2007. PMID: 17490627 Free PMC article. Review.
-
Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor.Cell. 2003 Oct 17;115(2):139-50. doi: 10.1016/s0092-8674(03)00803-1. Cell. 2003. PMID: 14567912 Free PMC article.
-
Hydrogen peroxide regulation of endothelial exocytosis by inhibition of N-ethylmaleimide sensitive factor.J Cell Biol. 2005 Jul 4;170(1):73-9. doi: 10.1083/jcb.200502031. J Cell Biol. 2005. PMID: 15998800 Free PMC article.
Cited by
-
N-ethylmaleimide‑sensitive factor siRNA inhibits the release of Weibel-Palade bodies in endothelial cells.Mol Med Rep. 2016 Aug;14(2):1061-6. doi: 10.3892/mmr.2016.5372. Epub 2016 Jun 7. Mol Med Rep. 2016. PMID: 27277949 Free PMC article.
-
Thioredoxin-mediated denitrosylation regulates cytokine-induced nuclear factor κB (NF-κB) activation.J Biol Chem. 2014 Jan 31;289(5):3066-72. doi: 10.1074/jbc.M113.503938. Epub 2013 Dec 12. J Biol Chem. 2014. PMID: 24338024 Free PMC article.
-
Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling.Antioxid Redox Signal. 2019 Apr 1;30(10):1331-1351. doi: 10.1089/ars.2017.7403. Epub 2018 Jan 10. Antioxid Redox Signal. 2019. PMID: 29130312 Free PMC article. Review.
-
Time-course investigation of SAGM-stored leukocyte-filtered red bood cell concentrates: from metabolism to proteomics.Haematologica. 2012 Jan;97(1):107-15. doi: 10.3324/haematol.2011.051789. Epub 2011 Oct 11. Haematologica. 2012. PMID: 21993682 Free PMC article.
-
Syntaxin-binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion.J Clin Invest. 2014 Oct;124(10):4503-16. doi: 10.1172/JCI71245. Epub 2014 Sep 17. J Clin Invest. 2014. PMID: 25244095 Free PMC article.
References
-
- Kubes P., McCafferty D. M. (2000) Am. J. Med. 109, 150–158 - PubMed
-
- Lefer A. M., Lefer D. J. (1999) Am. J. Physiol. 276, G572–575 - PubMed
-
- Grisham M. B., Jourd'Heuil D., Wink D. A. (1999) Am. J. Physiol. 276, G315–321 - PubMed
-
- Cirino G., Fiorucci S., Sessa W. C. (2003) Trends Pharmacol. Sci. 24, 91–95 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
