

doi: 10.1101/gad.1984311.
JNK regulates FoxO-dependent autophagy in neurons
Affiliations
- PMID: 21325132
- PMCID: PMC3042155
- DOI: 10.1101/gad.1984311
Item in Clipboard
JNK regulates FoxO-dependent autophagy in neurons
Genes Dev.
.
Abstract
The cJun N-terminal kinase (JNK) signal transduction pathway is implicated in the regulation of neuronal function. JNK is encoded by three genes that play partially redundant roles. Here we report the creation of mice with targeted ablation of all three Jnk genes in neurons. Compound JNK-deficient neurons are dependent on autophagy for survival. This autophagic response is caused by FoxO-induced expression of Bnip3 that displaces the autophagic effector Beclin-1 from inactive Bcl-XL complexes. These data identify JNK as a potent negative regulator of FoxO-dependent autophagy in neurons.
Figures
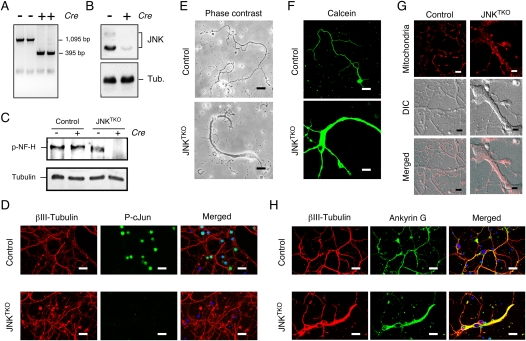
Establishment of JNK-deficient neurons. Wild-type (control) and Jnk1LoxP/LoxP Jnk2−/− Jnk3−/− CGNs were infected with Ad-cre at 3 d of culture in vitro (DIV) and then examined at 10 DIV. (A) Genotype analysis of JNKTKO neurons. The floxed Jnk1 and deleted Jnk1 alleles are detected as 1095-base-pair (bp) and 395-bp PCR products, respectively. (B) Extracts prepared from JNKTKO neurons were examined by immunoblot analysis using antibodies to JNK and α-Tubulin. (C) Control and JNKTKO neurons were examined at 10 DIV by immunoblot analysis using antibodies to phospho-neurofilament H and α-Tubulin. (D) Control and JNKTKO neurons were examined by immunofluorescence microscopy by staining with DAPI and antibodies to βIII tubulin and phospho-Ser63 cJun. Bar, 20 μm. (E) Control and JNKTKO neurons were examined by phase-contrast microscopy. Bar, 75 μm. (F) Control and JNKTKO neurons were stained with calcein-am ester and examined by fluorescence microscopy. Bar, 65 μm. (G) Wild-type (control) and JNKTKO neurons were stained with Mitotracker Red at 10 DIV and imaged by differential interference contrast (DIC) and fluorescence microscopy. Bar, 8 μm. (H) Control and JNKTKO neurons were examined by immunofluorescence microscopy by staining with DAPI and antibodies to βIII tubulin and Ankyrin G. Bar, 20 μm.
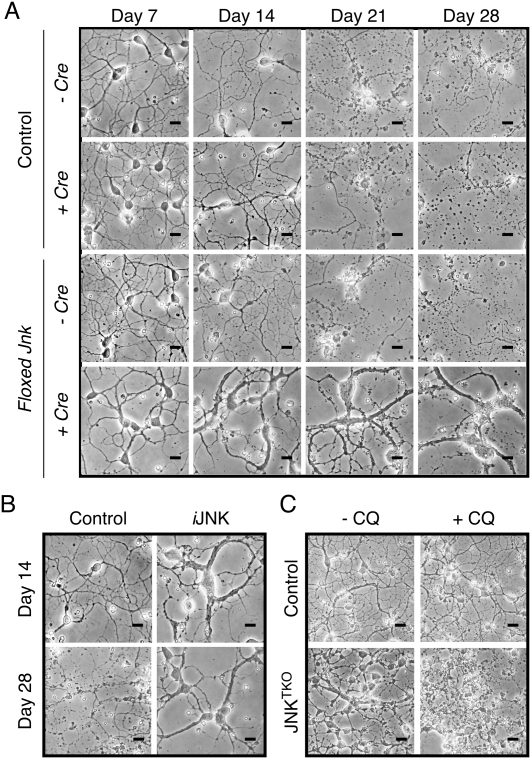
Increased life span of JNK-deficient neurons during culture in vitro. (A) Wild-type (control) and JNKTKO CGNs infected without or with Ad-cre at 3 DIV were examined by phase-contrast microscopy at 7–28 DIV. Bar, 45 μm. (B) Jnk1LoxP/LoxP Jnk2M108G/M108G Jnk3−/− CGNs were untreated (control) or infected with Ad-cre at 3 DIV and with the drug 1-NM-PP1 (1 μM) at 7 DIV (iJNK). The CGNs were examined using phase-contrast microscopy. Bar, 45 μm. (C) Wild-type (control) and JNKTKO CGNs infected with Ad-cre at 3 DIV were incubated without and with 1 μM chloroquine (CQ) at 11 DIV and then examined by phase-contrast microscopy at 12 DIV. Bar, 65 μm. Quantitative analysis of neuronal viability is presented in Supplemental Figure S3.
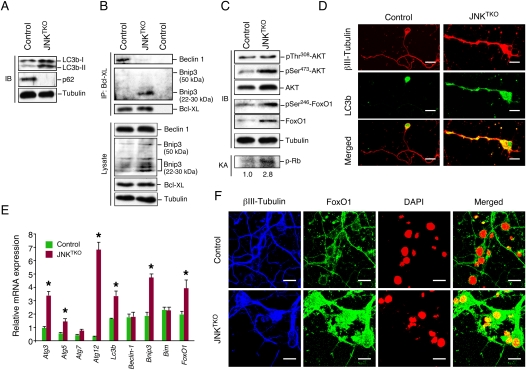
JNK deficiency in neurons causes increased autophagy. (A) Wild-type (control) and JNKTKO CGNs infected with Ad-cre at 3 DIV were harvested at 10 DIV to prepare protein extracts that were examined using antibodies to LC3b, p62/SQSTM1, and α-Tubulin. (B) Extracts prepared from control and JNKTKO CGNs were examined by immunoblot analysis by probing with antibodies to Bcl-XL, Bnip3, Beclin-1, and α-Tubulin. Coimmunoprecipitation assays were performed by immunoblot analysis of Bcl-XL immunoprecipitates. (C) Extracts prepared from control and JNKTKO CGNs were examined by immunoblot (IB) analysis by probing with antibodies to AKT, pSer308-AKT, pSer473-AKT, FoxO1, pSer246-FoxO1, and α-Tubulin. CDK2 activity was measured in an immunecomplex kinase assay (KA) using Rb as the substrate. The relative CDK2 activity is indicated below. (D) Control and JNKTKO CGNs were stained with βIII-Tubulin and LC3b antibodies and examined by fluorescence microscopy. Bar, 10 μm. (E) Gene expression in CGNs was examined by quantitative RT–PCR analysis of mRNA and normalized to the amount of Gapdh mRNA in each sample (mean ± SD; n = 3). Statistically significant differences are indicated. (*) P < 0.05. (F) Control and JNKTKO CGNs were stained with DAPI and antibodies to FoxO1 and βIII-Tubulin. The neurons were examined by fluorescence microscopy. The merged image represents colocalization of FoxO1 with DAPI. Bar, 10 μm.
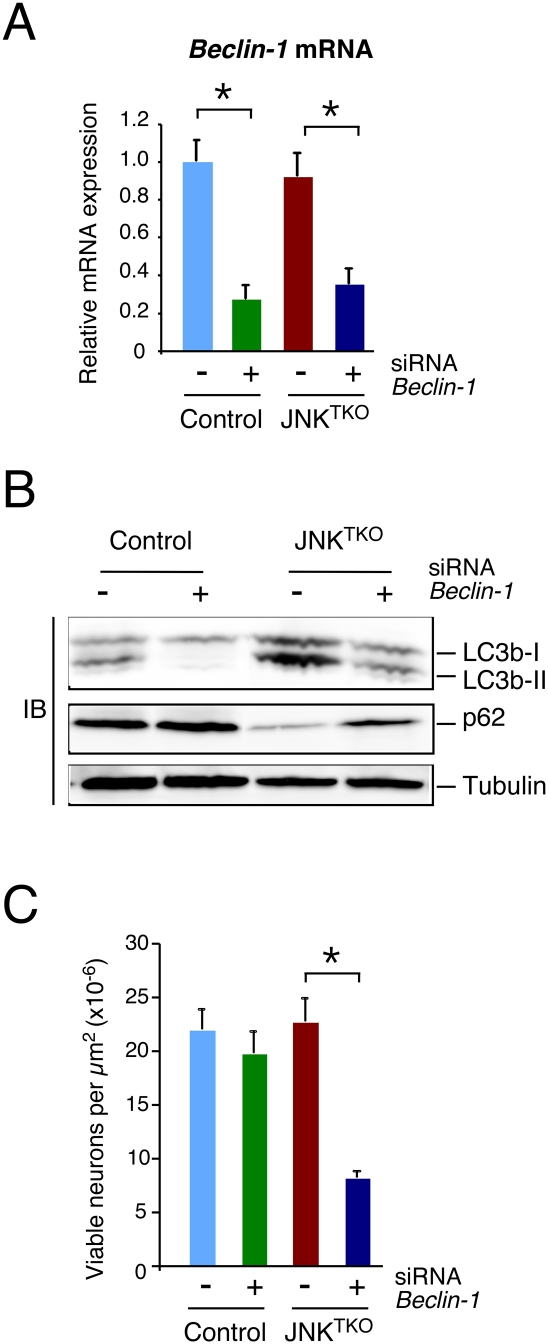
Effect of RNAi-mediated knockdown of Beclin-1 on autophagy and survival of JNKTKO neurons. (A) Wild-type (control) and Jnk1LoxP/LoxP Jnk2−/− Jnk3−/− (JNKTKO) neurons infected with Ad-cre at 3 DIV were transfected at 7 DIV with Beclin-1 siRNA or control siRNA. The expression of Beclin-1 mRNA was examined at 11 DIV by quantitative RT–PCR analysis of mRNA and normalized to the amount of Gapdh mRNA in each sample (mean ± SD; n = 3). Statistically significant differences are indicated. (*) P < 0.05. (B) Control and JNKTKO neurons transfected with scrambled sequence or Beclin-1 siRNA were examined at 11 DIV by immunoblot analysis with antibodies to LC3b, p62/SQSTM1, and α-Tubulin. (C) The survival of RNAi transfected control and JNKTKO neurons at 11 DIV was quantitated (mean ± SD; n = 20). Statistically significant differences are indicated. (*) P < 0.05.
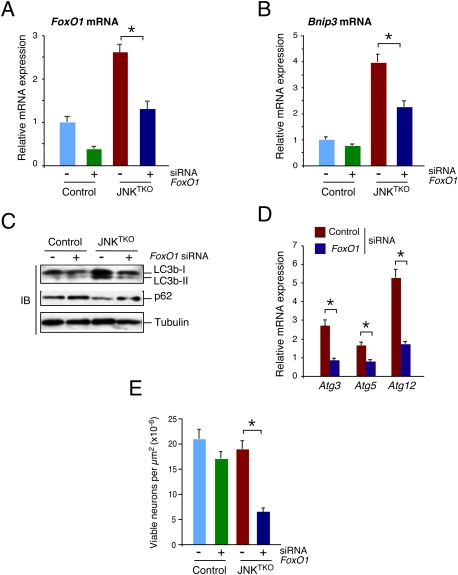
Effect of RNAi-mediated knockdown of FoxO1 on autophagy and survival of JNKTKO neurons. (A,B) Wild-type (control) and Jnk1LoxP/LoxP Jnk2−/− Jnk3−/− (JNKTKO) neurons infected with Ad-cre at 3 DIV were transfected at 7 DIV with FoxO1 siRNA or control siRNA. The expression of FoxO1 mRNA (A) and Bnip3 mRNA (B) was examined at 11 DIV by quantitative RT–PCR analysis of mRNA and normalized to the amount of Gapdh mRNA in each sample (mean ± SD; n = 3). Statistically significant differences are indicated. (*) P < 0.05. (C) Control and JNKTKO neurons transfected with scrambled sequence or FoxO1 siRNA were examined at 11 DIV by immunoblot analysis with antibodies to LC3b, p62/SQSTM1, and α-Tubulin. (D) RNAi transfected JNKTKO neurons were examined at 11 DIV by quantitative RT–PCR analysis of Atg3, Atg5, and Atg12 mRNA and normalized to the amount of Gapdh mRNA in each sample (mean ± SD; n = 3). Statistically significant differences are indicated. (*) P < 0.05. (E) The survival of RNAi transfected control and JNKTKO neurons at 11 DIV was quantitated (mean ± SD; n = 20). Statistically significant differences are indicated. (*) P < 0.05.
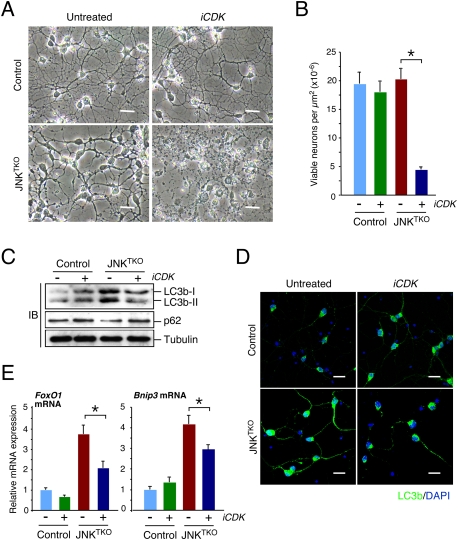
CDK activity is required for the viability of JNKTKO neurons. (A,B) Wild-type (control) and Jnk1f/f Jnk2−/− Jnk3−/− (JNKTKO) neurons infected with Ad-cre at 3 DIV were treated without or with the CDK inhibitor roscovitine (5 μM; iCDK) at 10 DIV. (A) The neurons were examined by phase-contrast microscopy at 11 DIV. Bar, 45 μm. (B) The number of viable neurons was examined at 11 DIV (mean ± SD; n = 20). Statistically significant differences are indicated. (*) P < 0.05. (C) Control and JNKTKO neurons were examined after treatment on 10 DIV with roscovitine (iCDK) for 8 h by immunoblot analysis using antibodies to LC3b, p62SQTM1, and α-Tubulin. (D) Control and JNKTKO neurons were examined by immunofluorescence analysis after treatment with roscovitine (iCDK) for 8 h using an antibody to LC3b. DNA was stained with DAPI. Bar, 20 μm. (E) Control and JNKTKO neurons were examined after treatment with roscovitine (iCDK) for 8 h by quantitative RT–PCR analysis of FoxO1 and Bnip3 mRNA and normalized to the amount of Gapdh mRNA in each sample (mean ± SD; n = 3). Statistically significant differences are indicated. (*) P < 0.05.
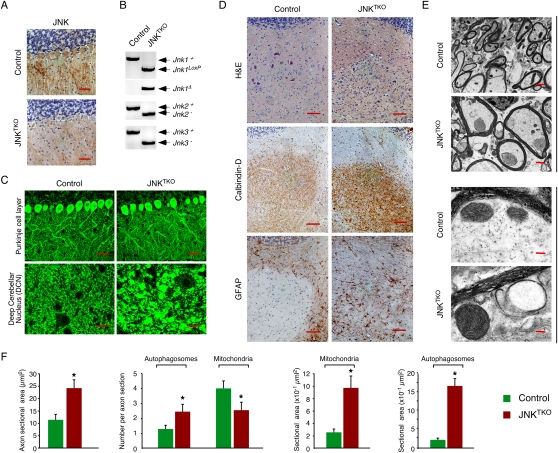
Compound deficiency of JNK in neurons in vivo. Young adult (8-wk-old) Pcp2-Cre mice (control) and Pcp2-Cre Jnk1LoxP/LoxP Jnk2−/− Jnk3−/− mice (JNKTKO) that express Cre recombinase selectively in cerebellar Purkinje cells were examined. (A) Sections of the Purkinje cell layer of control and JNKTKO mice were examined by immunohistochemical staining with antibodies to JNK1/2. Bar, 100 μm. (B) Cerebellar DNA was examined by PCR analysis to detect Jnk1+ (1550-bp), Jnk1LoxP (1095-bp), Jnk1Δ (395-bp), Jnk2+ (400-bp), Jnk2− (270-bp), Jnk3+ (430-bp), and Jnk3− (250-bp) alleles. (C) Sections of the Purkinje cell layer and DCN of control and JNKTKO mice were examined by immunofluorescence staining with an antibody to Calbindin D-28k. Bar, 40 μm. (D) Serial sections of the DCN of control and JNKTKO mice were examined by staining with H&E and by immunohistochemical staining antibodies to Calbindin D-28k and GFAP. Bar, 100 μm. (E) The myelinated axons in the DCN of control and JNKTKO mice were examined by transmission electron microscopy. Bars: top panels, 2 μm; bottom panels 0.125μm. (F) The axon area and the number and area of autophagosomes and mitochondria in the myelinated axons of control and JNKTKO mice were measured. The data are presented as mean ± SEM of 20 axons of three different mice per group. Statistically significant differences between control and JNKTKO mice are indicated. (*) P < 0.05.
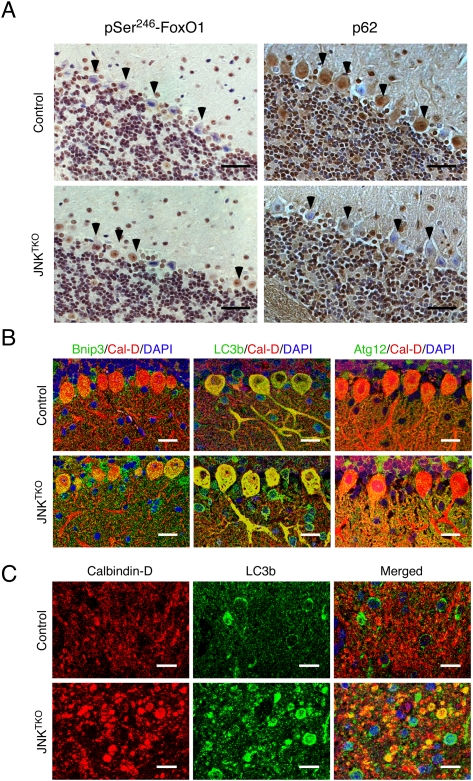
Compound JNK deficiency in Purkinje cells causes increased autophagy. Young adult (8-wk-old) Pcp2-Cre mice (control) and Pcp2-Cre Jnk1LoxP/LoxP Jnk2−/− Jnk3−/− mice (JNKTKO) that express Cre recombinase selectively in cerebellar Purkinje cells were examined. (A) Sections of the Purkinje cell layer of Pcp2-Cre mice (control) and Pcp2-Cre Jnk1LoxP/LoxP Jnk2−/− Jnk3−/− mice (JNKTKO) were examined by immunohistochemical staining with antibodies to pSer246-FoxO1 and p62/SQSTM1. Bar, 100 μm. (B) Sections of the Purkinje cell layer of control and JNKTKO mice were stained with antibodies (to Bnip3, LC3b, Atg12, and Calbidin-D28k) and examined by fluorescence microscopy. Bar, 20 μm. (C) Sections of the DCN of control and JNKTKO mice were stained with antibodies (to Calbindin D-28k and LC3b) and examined by fluorescence microscopy. Bar, 10 μm.
References
-
- Barski JJ, Dethleffsen K, Meyer M 2000. Cre recombinase expression in cerebellar Purkinje cells. Genesis 28: 93–98 - PubMed
-
- Bjorkblom B, Ostman N, Hongisto V, Komarovski V, Filen JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET 2005. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: Role of microtubule-associated protein 2 as an effector. J Neurosci 25: 6350–6361 - PMC - PubMed
-
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C 2003. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9: 1180–1186 - PubMed
-
- Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M, et al. 2005. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci 21: 363–377 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials