

Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited
- PMID: 21325631
- PMCID: PMC3078080
- DOI: 10.1091/mbc.E10-07-0599
Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited
Abstract
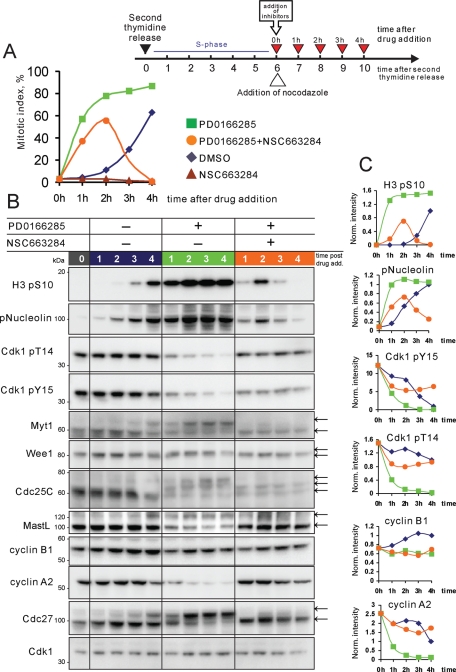
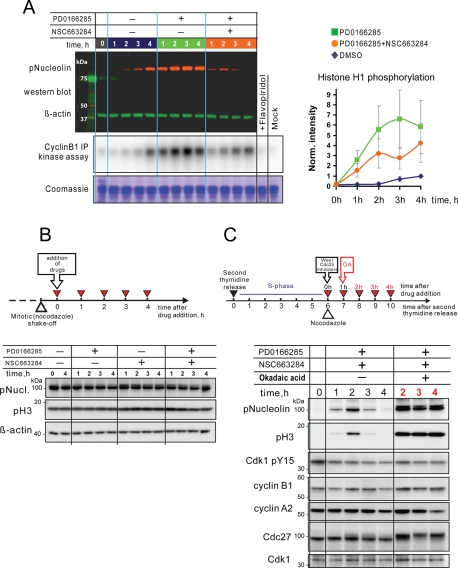
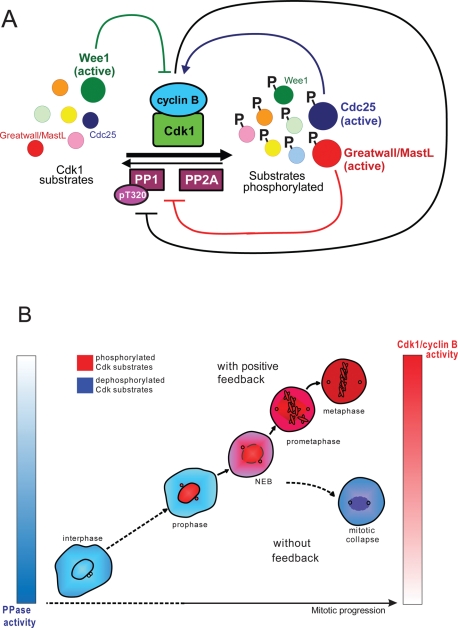
Mitosis requires precise coordination of multiple global reorganizations of the nucleus and cytoplasm. Cyclin-dependent kinase 1 (Cdk1) is the primary upstream kinase that directs mitotic progression by phosphorylation of a large number of substrate proteins. Cdk1 activation reaches the peak level due to positive feedback mechanisms. By inhibiting Cdk chemically, we showed that, in prometaphase, when Cdk1 substrates approach the peak of their phosphorylation, cells become capable of proper M-to-G1 transition. We interfered with the molecular components of the Cdk1-activating feedback system through use of chemical inhibitors of Wee1 and Myt1 kinases and Cdc25 phosphatases. Inhibition of Wee1 and Myt1 at the end of the S phase led to rapid Cdk1 activation and morphologically normal mitotic entry, even in the absence of G2. Dampening Cdc25 phosphatases simultaneously with Wee1 and Myt1 inhibition prevented Cdk1/cyclin B kinase activation and full substrate phosphorylation and induced a mitotic "collapse," a terminal state characterized by the dephosphorylation of mitotic substrates without cyclin B proteolysis. This was blocked by the PP1/PP2A phosphatase inhibitor, okadaic acid. These findings suggest that the positive feedback in Cdk activation serves to overcome the activity of Cdk-opposing phosphatases and thus sustains forward progression in mitosis.
Figures
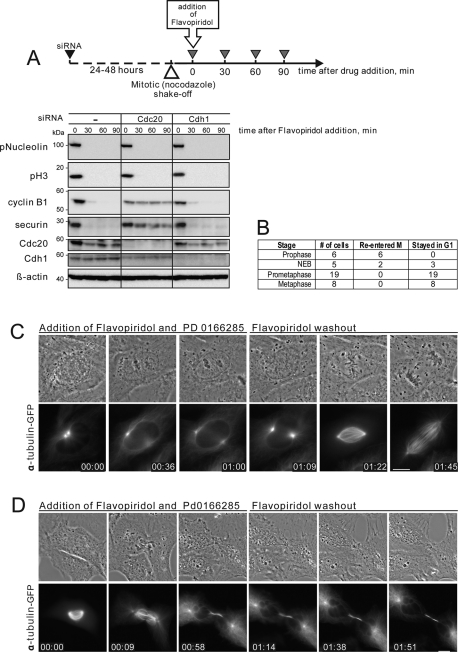
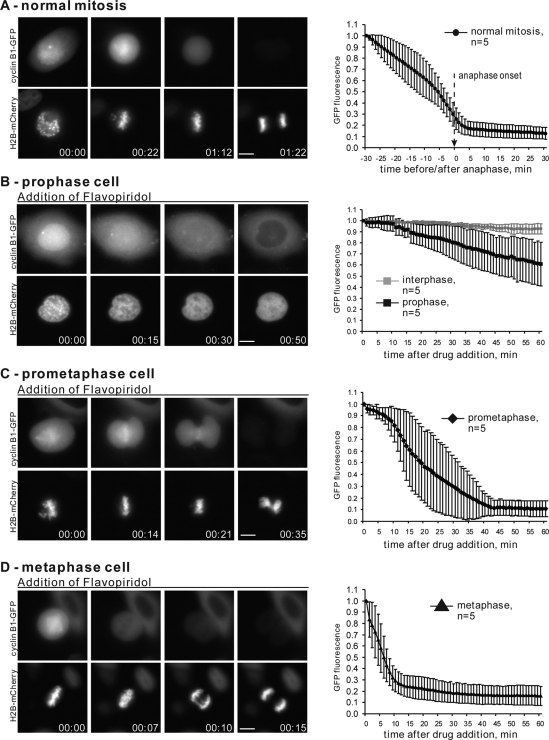
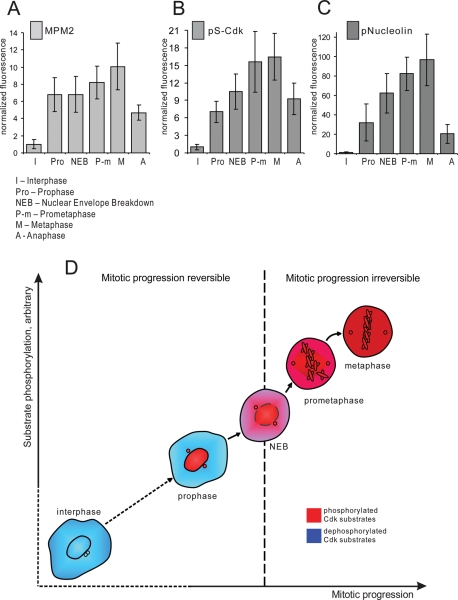
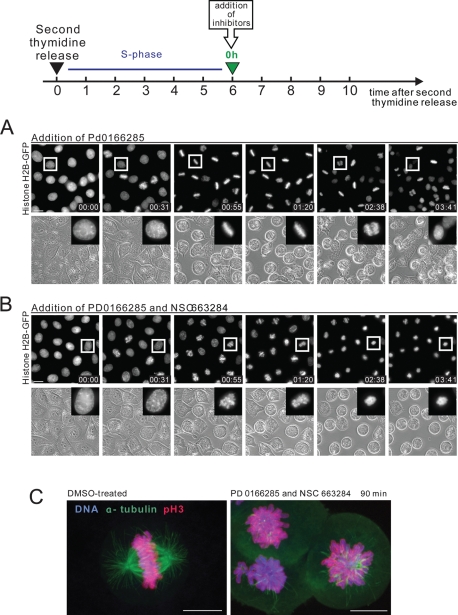
References
-
- Andreeva AV, Kutuzov MA. PPP family of protein Ser/Thr phosphatases: two distinct branches? Mol Biol Evol. 2001;18:448–452. - PubMed
-
- Arooz T, Yam CH, Siu WY, Lau A, Li KK, Poon RY. On the concentrations of cyclins and cyclin-dependent kinases in extracts of cultured human cells. Biochemistry. 2000;39:9494–9501. - PubMed
-
- Bellanger S, de Gramont A, Sobczak-Thepot J. Cyclin B2 suppresses mitotic failure and DNA rereplication in human somatic cells knocked down for both cyclins B1 and B2. Oncogene. 2007;26:7175–7184. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
