

De novo MECP2 duplication in two females with random X-inactivation and moderate mental retardation
- PMID: 21326285
- PMCID: PMC3083613
- DOI: 10.1038/ejhg.2010.226
De novo MECP2 duplication in two females with random X-inactivation and moderate mental retardation
Abstract
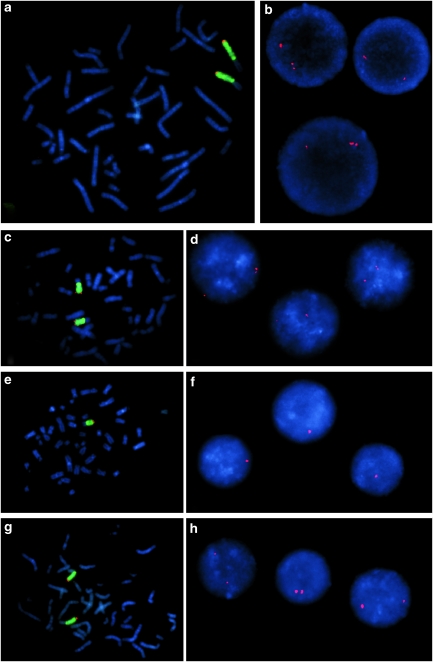
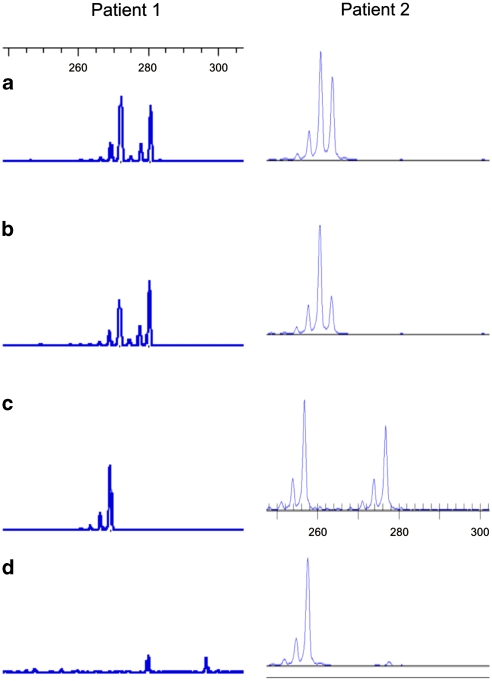
Xq28 duplications including MECP2 are a well-known cause of severe mental retardation in males with seizures, muscular hypotonia, progressive spasticity, poor speech and recurrent infections that often lead to early death. Female carriers usually show a normal intellectual performance due to skewed X-inactivation (XCI). We report on two female patients with a de novo MECP2 duplication associated with moderate mental retardation. In both patients, the de novo duplication occurred on the paternal allele, and both patients show a random XCI, which can be assumed as the triggering factor for the phenotype. Furthermore, we describe the phenotype that might be restricted to unspecific mild-to -moderate mental retardation with neurological features in early adulthood.
Figures

References
-
- Rett A. Ueber ein eigenartiges hirnatrophisches Syndrom bei Hyperammoniamie in Kindesalter. Wien Med Wschr. 1966;116:723–738. - PubMed
-
- Rett A.Cerebral atrophy associated with hyperammonaemiain Vinken PJ, Bruyn GW (eds): Handbook of Clinical Neurology Amsterdam: Elsevier; 1977. Vol 29, pp305–329.
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Lubs H, Abidi F, Bier JAB, et al. XLMR syndrome characterized by multiple respiratory infections, hypertelorism, severe CNS deterioration and early death localizes to distal Xq28. Am J Med Genet. 1999;85:243–248. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
