

Classic selective sweeps were rare in recent human evolution
- PMID: 21330547
- PMCID: PMC3669691
- DOI: 10.1126/science.1198878
Classic selective sweeps were rare in recent human evolution
Abstract
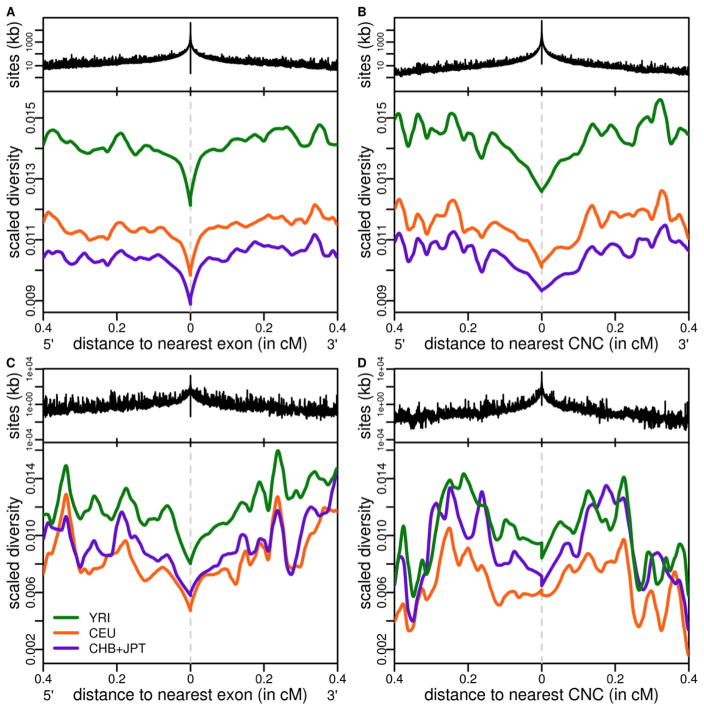
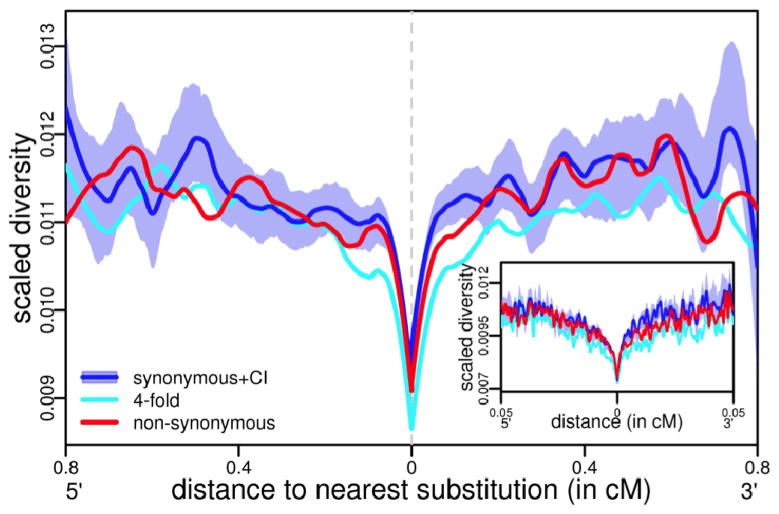
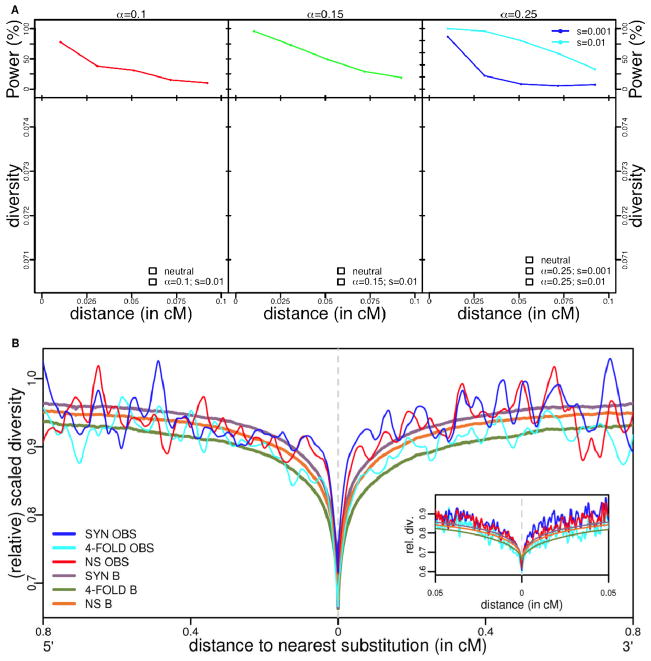
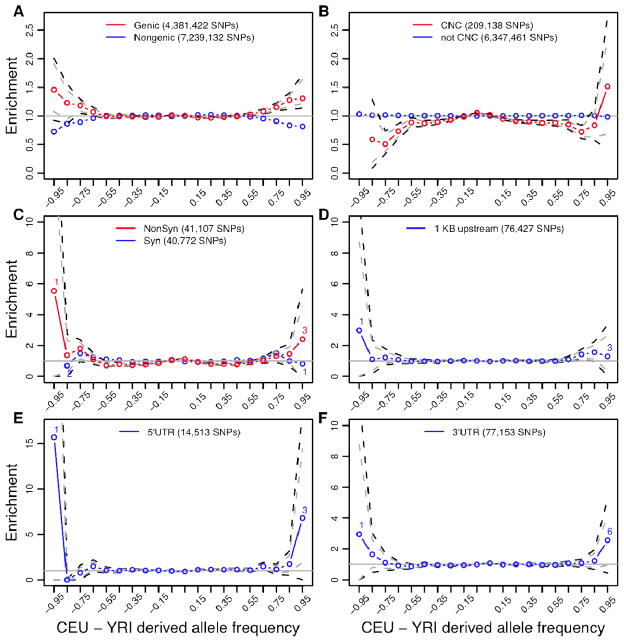
Efforts to identify the genetic basis of human adaptations from polymorphism data have sought footprints of "classic selective sweeps" (in which a beneficial mutation arises and rapidly fixes in the population).Yet it remains unknown whether this form of natural selection was common in our evolution. We examined the evidence for classic sweeps in resequencing data from 179 human genomes. As expected under a recurrent-sweep model, we found that diversity levels decrease near exons and conserved noncoding regions. In contrast to expectation, however, the trough in diversity around human-specific amino acid substitutions is no more pronounced than around synonymous substitutions. Moreover, relative to the genome background, amino acid and putative regulatory sites are not significantly enriched in alleles that are highly differentiated between populations. These findings indicate that classic sweeps were not a dominant mode of human adaptation over the past ~250,000 years.
Figures
Comment in
-
Human evolution: Sweep model is swept away.Nat Rev Genet. 2011 Apr;12(4):228-9. doi: 10.1038/nrg2978. Epub 2011 Mar 1. Nat Rev Genet. 2011. PMID: 21358742 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 GM072861/GM/NIGMS NIH HHS/United States
- GM087069/GM/NIGMS NIH HHS/United States
- GM083228/GM/NIGMS NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- R01 HG007644/HG/NHGRI NIH HHS/United States
- GM72861/GM/NIGMS NIH HHS/United States
- 086084/WT_/Wellcome Trust/United Kingdom
- F32 GM087069/GM/NIGMS NIH HHS/United States
- R01 GM083228/GM/NIGMS NIH HHS/United States
- WT086084MA/WT_/Wellcome Trust/United Kingdom
- HHMI/Howard Hughes Medical Institute/United States
- GM20373/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
