

Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2
- PMID: 21331077
- PMCID: PMC3172091
- DOI: 10.1038/cdd.2011.10
Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2
Abstract
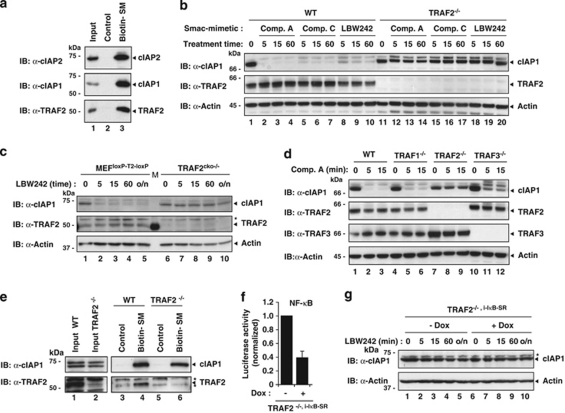
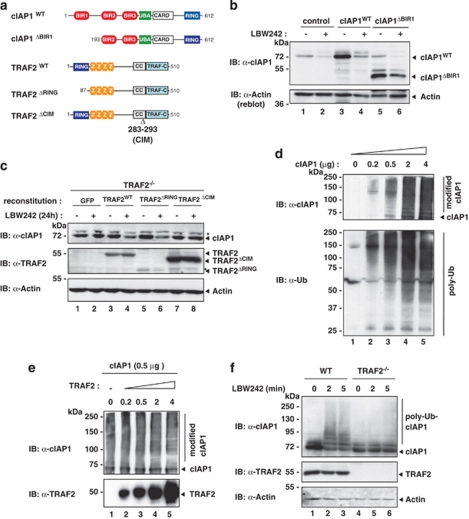
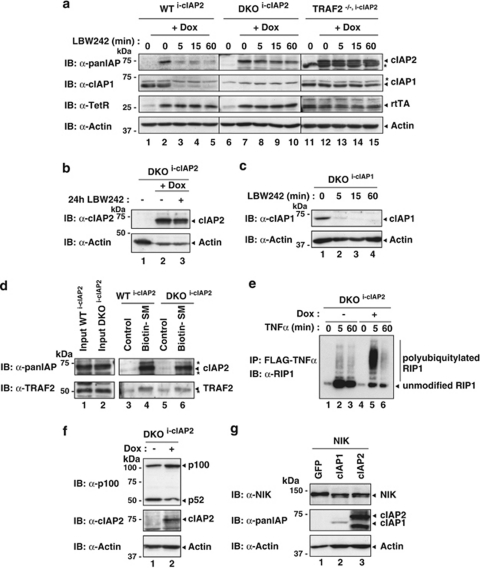
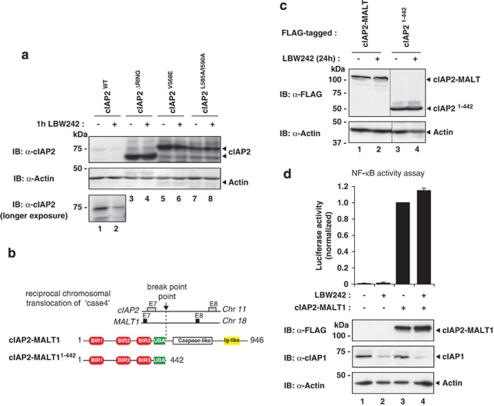
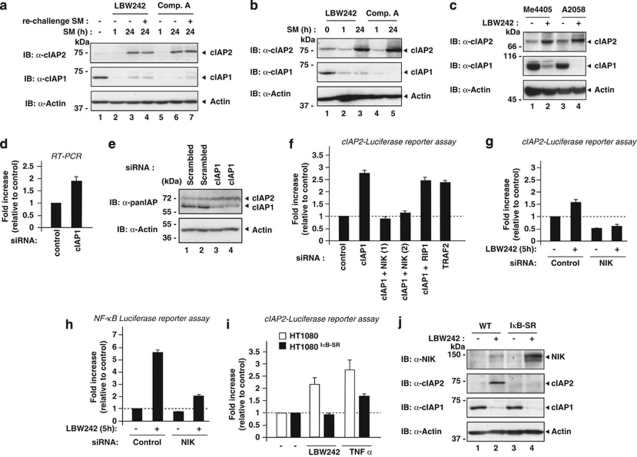
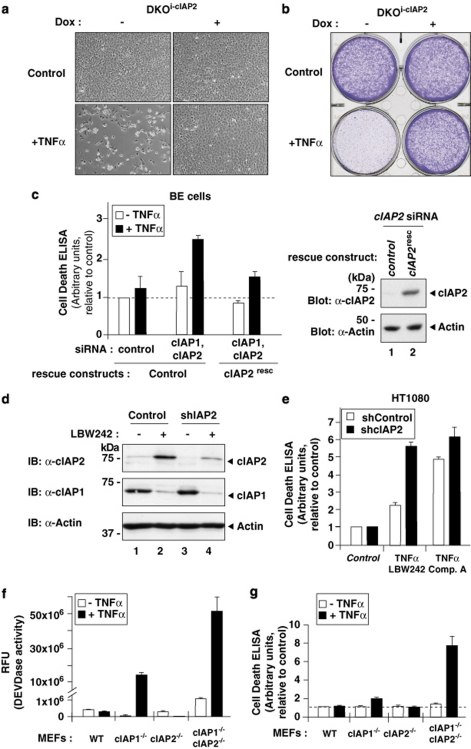
The inhibitors of apoptosis (IAP) proteins cIAP1 and cIAP2 have recently emerged as key ubiquitin-E3 ligases regulating innate immunity and cell survival. Much of our knowledge of these IAPs stems from studies using pharmacological inhibitors of IAPs, dubbed Smac mimetics (SMs). Although SMs stimulate auto-ubiquitylation and degradation of cIAPs, little is known about the molecular determinants through which SMs activate the E3 activities of cIAPs. In this study, we find that SM-induced rapid degradation of cIAPs requires binding to tumour necrosis factor (TNF) receptor-associated factor 2 (TRAF2). Moreover, our data reveal an unexpected difference between cIAP1 and cIAP2. Although SM-induced degradation of cIAP1 does not require cIAP2, degradation of cIAP2 critically depends on the presence of cIAP1. In addition, degradation of cIAP2 also requires the ability of the cIAP2 RING finger to dimerise and to bind to E2s. This has important implications because SM-mediated degradation of cIAP1 causes non-canonical activation of NF-κB, which results in the induction of cIAP2 gene expression. In the absence of cIAP1, de novo synthesised cIAP2 is resistant to the SM and suppresses TNFα killing. Furthermore, the cIAP2-MALT1 oncogene, which lacks cIAP2's RING, is resistant to SM treatment. The identification of mechanisms through which cancer cells resist SM treatment will help to improve combination therapies aimed at enhancing treatment response.
Figures
References
-
- Chen DJ, Huerta S. Smac mimetics as new cancer therapeutics. Anticancer Drugs. 2009;20:646–658. - PubMed
-
- Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun. 2003;304:499–504. - PubMed
-
- Cossu F, Mastrangelo E, Milani M, Sorrentino G, Lecis D, Delia D, et al. Designing Smac-mimetics as antagonists of XIAP, cIAP1, and cIAP2. Biochem Biophys Res Commun. 2009;378:162–167. - PubMed
-
- Yang YL, Li XM. The IAP family: endogenous caspase inhibitors with multiple biological activities. Cell Res. 2000;10:169–177. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
