

Adenosine kinase regulation of cardiomyocyte hypertrophy
- PMID: 21335462
- PMCID: PMC3094084
- DOI: 10.1152/ajpheart.00684.2010
Adenosine kinase regulation of cardiomyocyte hypertrophy
Abstract
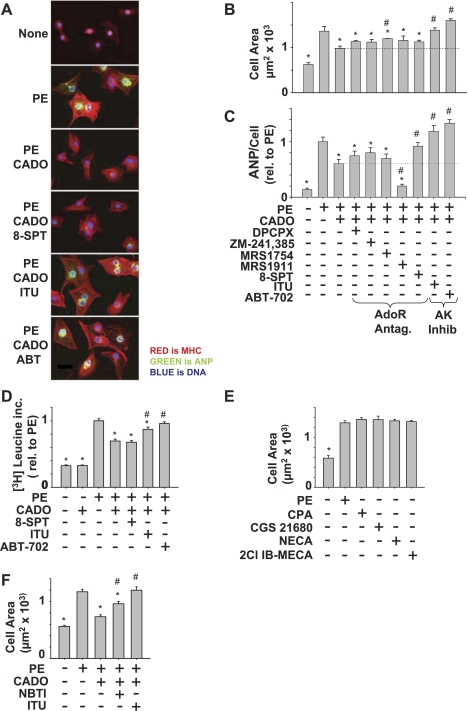
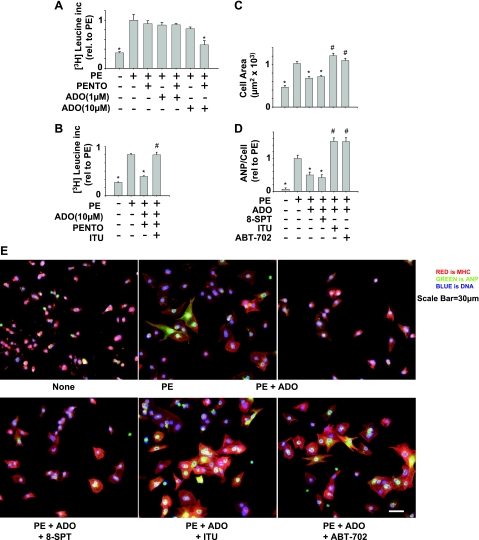
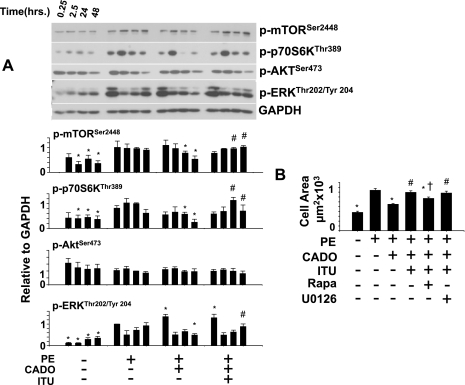
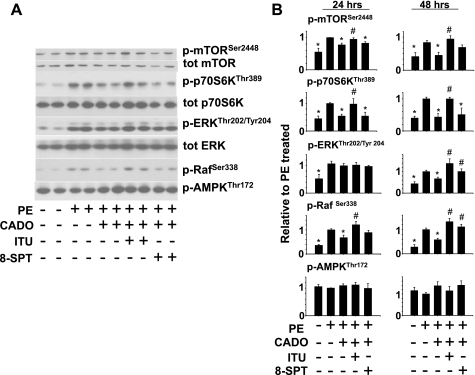
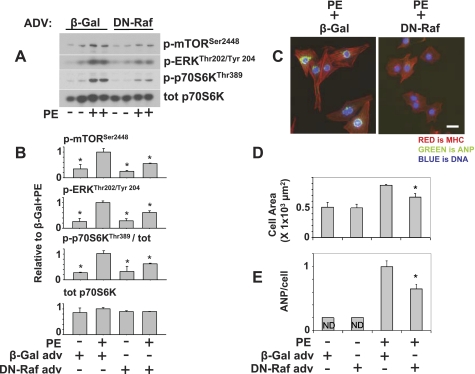
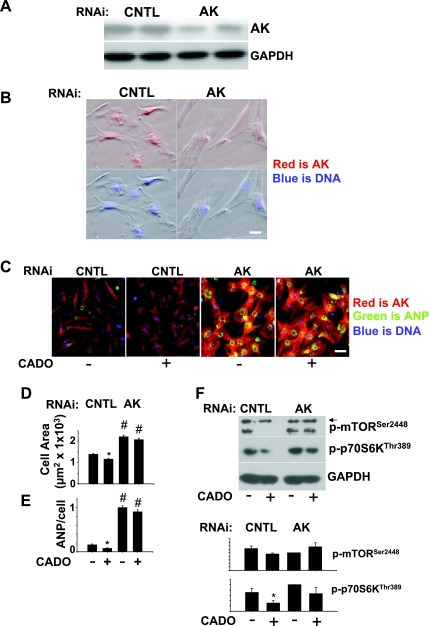
There is evidence that extracellular adenosine can attenuate cardiac hypertrophy, but the mechanism by which this occurs is not clear. Here we investigated the role of adenosine receptors and adenosine metabolism in attenuation of cardiomyocyte hypertrophy. Phenylephrine (PE) caused hypertrophy of neonatal rat cardiomyocytes with increases of cell surface area, protein synthesis, and atrial natriuretic peptide (ANP) expression. These responses were attenuated by 5 μM 2-chloroadenosine (CADO; adenosine deaminase resistant adenosine analog) or 10 μM adenosine. While antagonism of adenosine receptors partially blocked the reduction of ANP expression produced by CADO, it did not restore cell size or protein synthesis. In support of a role for intracellular adenosine metabolism in regulating hypertrophy, the adenosine kinase (AK) inhibitors iodotubercidin and ABT-702 completely reversed the attenuation of cell size, protein synthesis, and expression of ANP by CADO or ADO. Examination of PE-induced phosphosignaling pathways revealed that CADO treatment did not reduce AKT(Ser⁴⁷³) phosphorylation but did attenuate sustained phosphorylation of Raf(Ser³³⁸) (24-48 h), mTOR(Ser²⁴⁴⁸) (24-48 h), p70S6k(Thr³⁸⁹) (2.5-48 h), and ERK(Thr²⁰²/Tyr²⁰⁴) (48 h). Inhibition of AK restored activation of these enzymes in the presence of CADO. Using dominant negative and constitutively active Raf adenoviruses, we found that Raf activation is necessary and sufficient for PE-induced mTORC1 signaling and cardiomyocyte hypertrophy. CADO treatment still blocked p70S6k(Thr³⁸⁹) phosphorylation and hypertrophy downstream of constitutively active Raf, however, despite a high level phosphorylation of ERK(Thr202/Tyr204) and AKT(Ser⁴⁷³). Reduction of Raf-induced p70S6k(Thr³⁸⁹) phosphorylation and hypertrophy by CADO was reversed by inhibiting AK. Together, these results identify AK as an important mediator of adenosine attenuation of cardiomyocyte hypertrophy, which acts, at least in part, through inhibition of Raf signaling to mTOR/p70S6k.
Figures

Similar articles
-
Adenosine kinase attenuates cardiomyocyte microtubule stabilization and protects against pressure overload-induced hypertrophy and LV dysfunction.J Mol Cell Cardiol. 2019 May;130:49-58. doi: 10.1016/j.yjmcc.2019.03.015. Epub 2019 Mar 22. J Mol Cell Cardiol. 2019. PMID: 30910669 Free PMC article.
-
ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2, 3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties: I. In vitro characterization and acute antinociceptive effects in the mouse.J Pharmacol Exp Ther. 2000 Dec;295(3):1156-64. J Pharmacol Exp Ther. 2000. PMID: 11082453
-
Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway.J Endocrinol. 2009 Aug;202(2):299-307. doi: 10.1677/JOE-09-0044. Epub 2009 May 27. J Endocrinol. 2009. PMID: 19474060
-
Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways.J Biol Chem. 2011 May 27;286(21):18465-73. doi: 10.1074/jbc.M110.200022. Epub 2011 Apr 8. J Biol Chem. 2011. PMID: 21478152 Free PMC article.
-
Indoleamine 2,3-Dioxygenase 1 (IDO1) Promotes Cardiac Hypertrophy via a PI3K-AKT-mTOR-Dependent Mechanism.Cardiovasc Toxicol. 2021 Aug;21(8):655-668. doi: 10.1007/s12012-021-09657-y. Epub 2021 May 21. Cardiovasc Toxicol. 2021. Retraction in: Cardiovasc Toxicol. 2023 Feb;23(2):120. doi: 10.1007/s12012-023-09779-5. PMID: 34021461 Free PMC article. Retracted.
Cited by
-
Role of adenosine in status epilepticus: a potential new target?Epilepsia. 2013 Sep;54 Suppl 6(0 6):20-2. doi: 10.1111/epi.12268. Epilepsia. 2013. PMID: 24001064 Free PMC article. Review.
-
Cardiac purinergic signalling in health and disease.Purinergic Signal. 2015 Mar;11(1):1-46. doi: 10.1007/s11302-014-9436-1. Epub 2014 Dec 20. Purinergic Signal. 2015. PMID: 25527177 Free PMC article. Review.
-
Pharmacological Tuning of Adenosine Signal Nuances Underlying Heart Failure With Preserved Ejection Fraction.Front Pharmacol. 2021 Aug 20;12:724320. doi: 10.3389/fphar.2021.724320. eCollection 2021. Front Pharmacol. 2021. PMID: 34489711 Free PMC article. Review.
-
Adenosine kinase attenuates cardiomyocyte microtubule stabilization and protects against pressure overload-induced hypertrophy and LV dysfunction.J Mol Cell Cardiol. 2019 May;130:49-58. doi: 10.1016/j.yjmcc.2019.03.015. Epub 2019 Mar 22. J Mol Cell Cardiol. 2019. PMID: 30910669 Free PMC article.
-
Adenosine kinase: exploitation for therapeutic gain.Pharmacol Rev. 2013 Apr 16;65(3):906-43. doi: 10.1124/pr.112.006361. Print 2013 Jul. Pharmacol Rev. 2013. PMID: 23592612 Free PMC article. Review.
References
-
- Aoki H, Sadoshima J, Izumo S. Myosin light chain kinase mediates sarcomere organization during cardiac hypertrophy in vitro. Nat Med 6: 183– 188, 2000 - PubMed
-
- Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem 279: 32771– 32779, 2004 - PubMed
-
- Cohen MV, Downey JM. Adenosine: trigger and mediator of cardioprotection. Basic Res Cardiol 103: 203– 215, 2008 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous