

Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer
- PMID: 21336312
- PMCID: PMC3918344
- DOI: 10.1038/ncb2170
Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer
Abstract
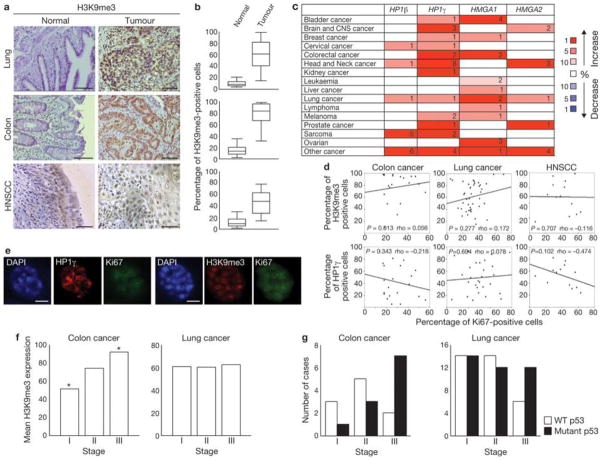
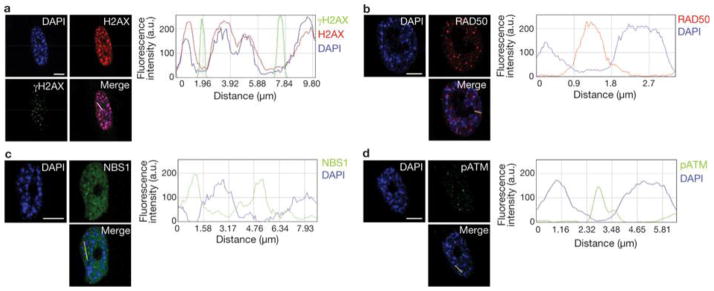
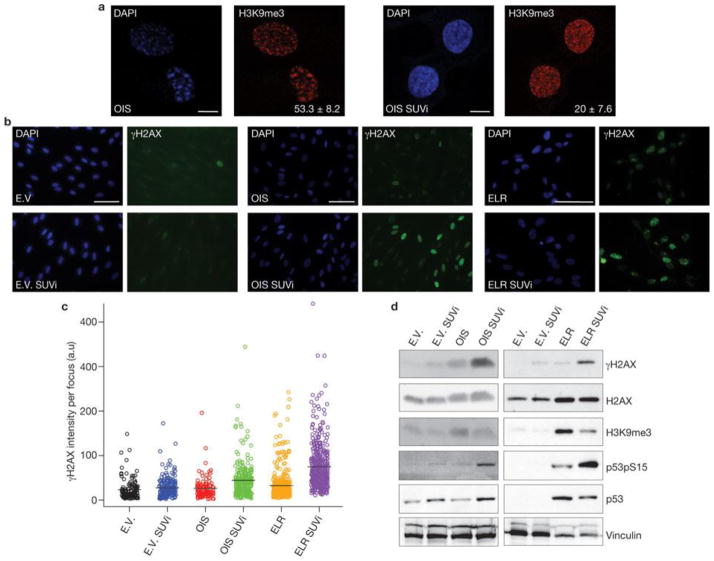
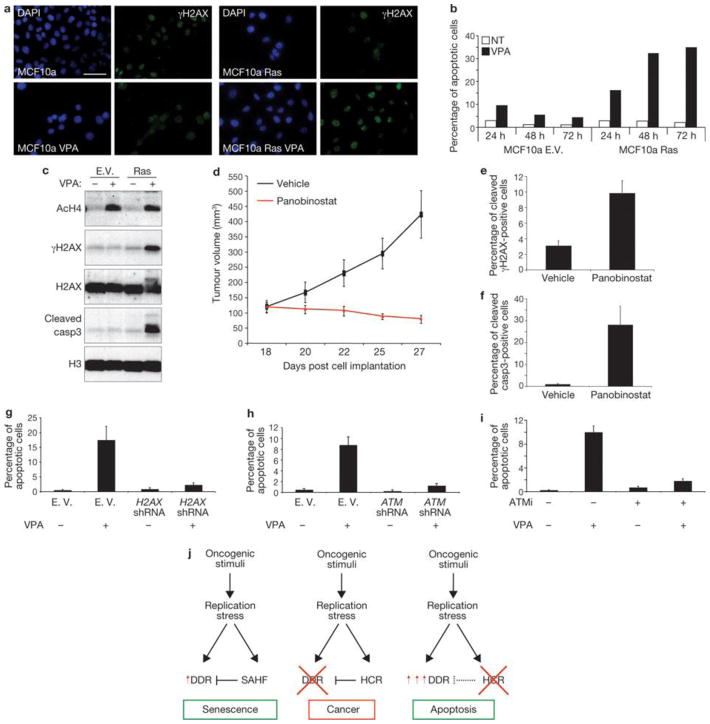
Two major mechanisms have been causally implicated in the establishment of cellular senescence: the activation of the DNA damage response (DDR) pathway and the formation of senescence-associated heterochromatic foci (SAHF). Here we show that in human fibroblasts resistant to premature p16(INK4a) induction, SAHF are preferentially formed following oncogene activation but are not detected during replicative cellular senescence or on exposure to a variety of senescence-inducing stimuli. Oncogene-induced SAHF formation depends on DNA replication and ATR (ataxia telangiectasia and Rad3-related). Inactivation of ATM (ataxia telangiectasia mutated) or p53 allows the proliferation of oncogene-expressing cells that retain increased heterochromatin induction. In human cancers, levels of heterochromatin markers are higher than in normal tissues, and are independent of the proliferative index or stage of the tumours. Pharmacological and genetic perturbation of heterochromatin in oncogene-expressing cells increase DDR signalling and lead to apoptosis. In vivo, a histone deacetylase inhibitor (HDACi) causes heterochromatin relaxation, increased DDR, apoptosis and tumour regression. These results indicate that heterochromatin induced by oncogenic stress restrains DDR and suggest that the use of chromatin-modifying drugs in cancer therapies may benefit from the study of chromatin and DDR status of tumours.
© 2011 Macmillan Publishers Limited. All rights reserved
Conflict of interest statement
S.M. has stocks in Genextra Spa, a biopharmaceutical company that is currently developing HDAC inhibitors for cancer therapy.
Figures
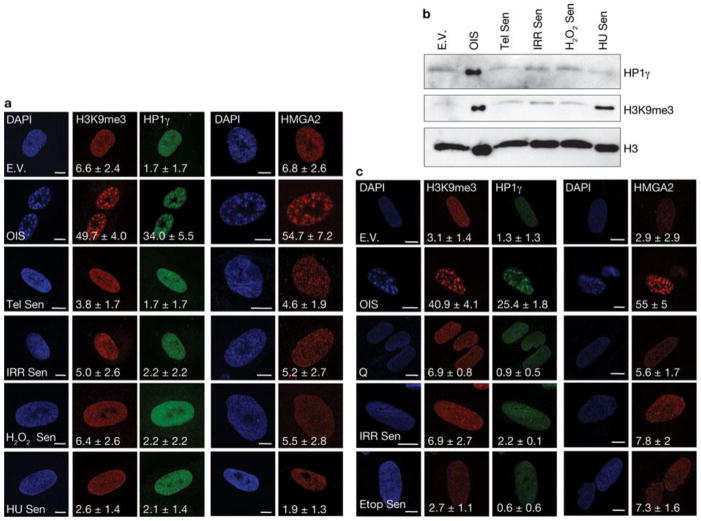
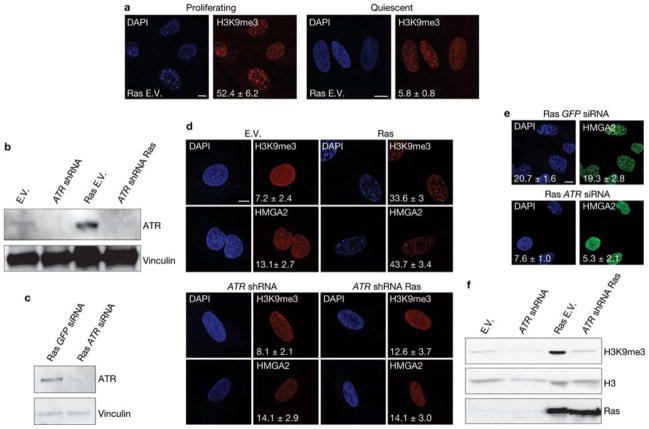
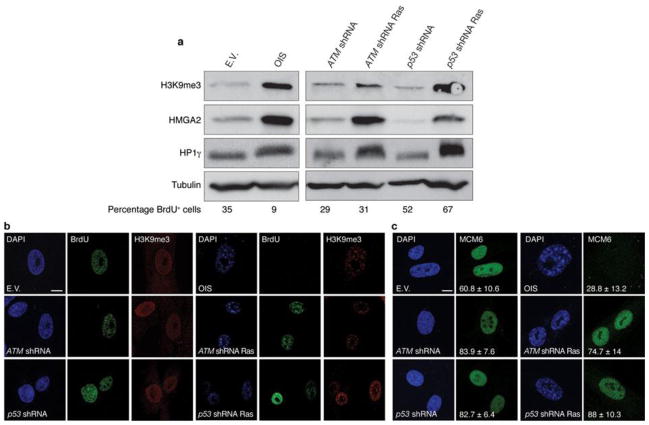
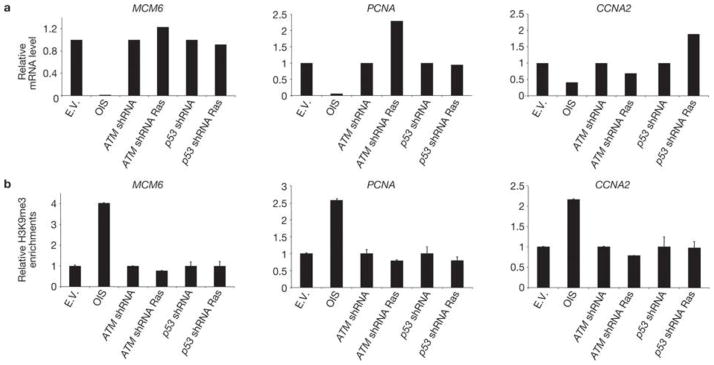
Comment in
-
A damage limitation exercise.Nat Cell Biol. 2011 Mar;13(3):193-5. doi: 10.1038/ncb0311-193. Nat Cell Biol. 2011. PMID: 21364567
References
-
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. - PubMed
-
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. - PubMed
-
- Adams PD. Healing and hurting: molecular mechanisms, functions and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. - PubMed
-
- d’Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. - PubMed
-
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
