

Does mitochondrial DNA play a role in Parkinson's disease? A review of cybrid and other supportive evidence
- PMID: 21338319
- PMCID: PMC3643260
- DOI: 10.1089/ars.2011.3948
Does mitochondrial DNA play a role in Parkinson's disease? A review of cybrid and other supportive evidence
Abstract
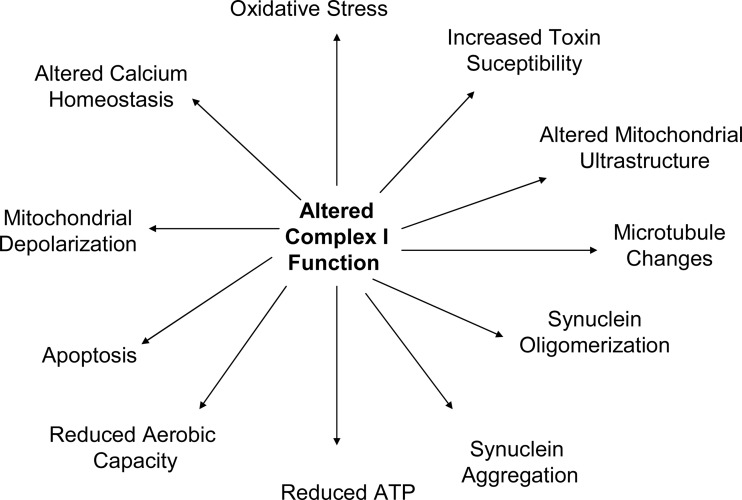
Significance: Mitochondria are currently believed to play an important role in the neurodysfunction and neurodegeneration that underlie Parkinson's disease (PD).
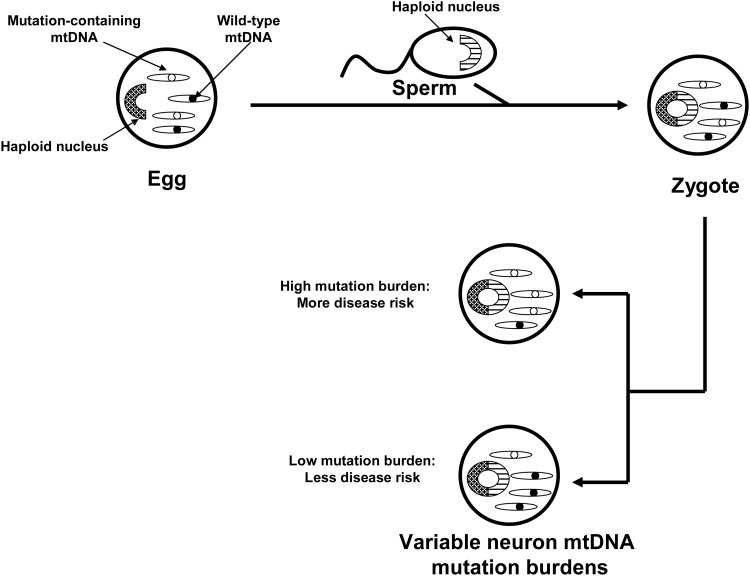
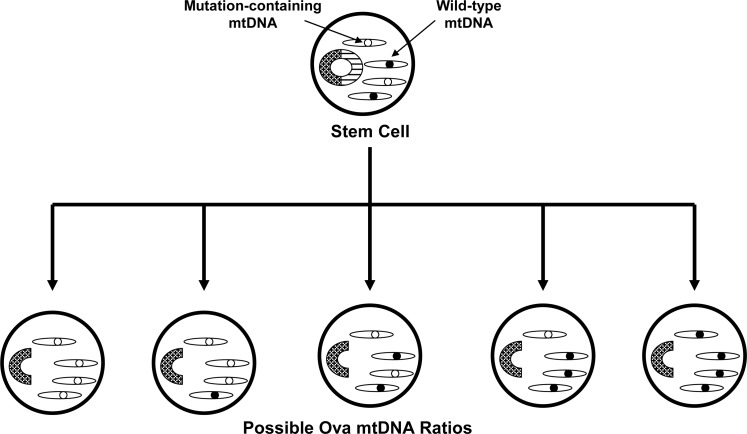
Recent advances: While it increasingly appears that mitochondrial dysfunction in PD can have different causes, it has been proposed that mitochondrial DNA (mtDNA) may account for or drive mitochondrial dysfunction in the majority of the cases. If correct, the responsible mtDNA signatures could represent acquired mutations, inherited mutations, or population-distributed polymorphisms.
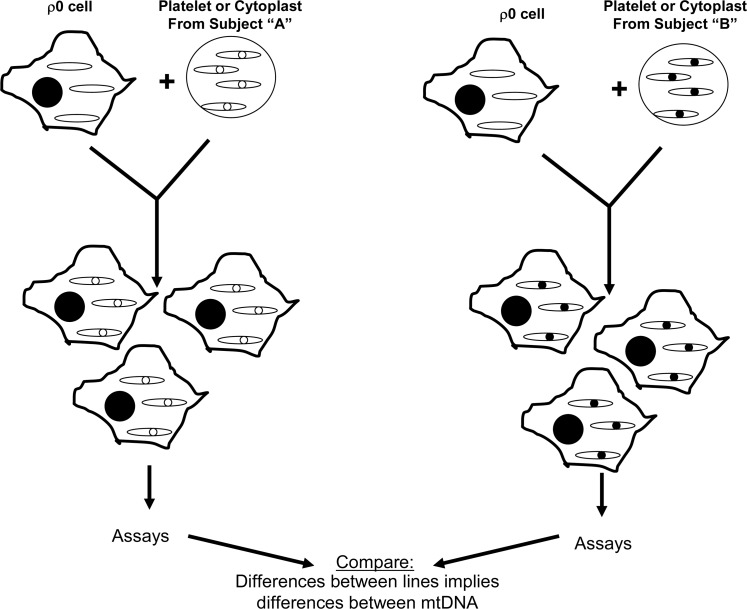
Critical issues and future directions: This review discusses the case for mtDNA as a key mediator of PD, and especially focuses on data from studies of PD cytoplasmic hybrid (cybrid) cell lines.
Figures
References
-
- Anderson S. Bankier AT. Barrell BG. de Bruijn MH. Coulson AR. Drouin J. Eperon IC. Nierlich DP. Roe BA. Sanger F. Schreier PH. Smith AJ. Staden R. Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
-
- Andrews RM. Kubacka I. Chinnery PF. Lightowlers RN. Turnbull DM. Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. - PubMed
-
- Aomi Y. Chen CS. Nakada K. Ito S. Isobe K. Murakami H. Kuno SY. Tawata M. Matsuoka R. Mizusawa H. Hayashi JI. Cytoplasmic transfer of platelet mtDNA from elderly patients with Parkinson's disease to mtDNA-less HeLa cells restores complete mitochondrial respiratory function. Biochem Biophys Res Commun. 2001;280:265–273. - PubMed
-
- Arthur CR. Morton SL. Dunham LD. Keeney PM. Bennett JP., Jr. Parkinson's disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol Neurodegener. 2009;4:37. - PMC - PubMed
-
- Autere J. Moilanen JS. Finnila S. Soininen H. Mannermaa A. Hartikainen P. Hallikainen M. Majamaa K. Mitochondrial DNA polymorphisms as risk factors for Parkinson's disease and Parkinson's disease dementia. Hum Genet. 2004;115:29–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
