

Biomarkers in rare disorders: the experience with spinal muscular atrophy
- PMID: 21339974
- PMCID: PMC3039940
- DOI: 10.3390/ijms12010024
Biomarkers in rare disorders: the experience with spinal muscular atrophy
Abstract
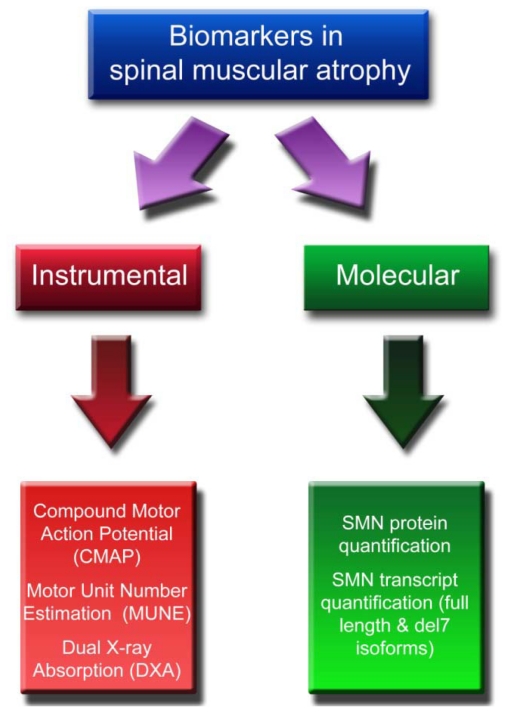
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder caused by homozygous mutations of the SMN1 gene. Based on clinical severity, three forms of SMA are recognized (type I-III). All patients have at least one (usually 2-4) copies of a highly homologous gene (SMN2) which produces insufficient levels of functional SMN protein, due to alternative splicing of exon7. Recently, evidence has been provided that SMN2 expression can be enhanced by different strategies. The availability of potential candidates to treat SMA has raised a number of issues, including the availability of data on the natural history of the disease, the reliability and sensitivity of outcome measures, the duration of the studies, and the number and clinical homogeneity of participating patients. Equally critical is the availability of reliable biomarkers. So far, different tools have been proposed as biomarkers in SMA, classifiable into two groups: instrumental (the Compound Motor Action Potential, the Motor Unit Number Estimation, and the Dual-energy X-ray absorptiometry) and molecular (SMN gene products dosage, either transcripts or protein). However, none of the biomarkers available so far can be considered the gold standard. Preclinical studies on SMA animal models and double-blind, placebo-controlled studies are crucial to evaluate the appropriateness of biomarkers, on the basis of correlations with clinical outcome.
Keywords: SMA; SMN; biomarker; spinal muscular atrophy.
Figures
References
-
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2033. - PubMed
-
- Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromuscul. Disord. 1995;5:3–5. - PubMed
-
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum. Mut. 2000;15:228–237. - PubMed
-
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
