

Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin
- PMID: 21339984
- PMCID: PMC3039950
- DOI: 10.3390/ijms12010226
Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin
Abstract
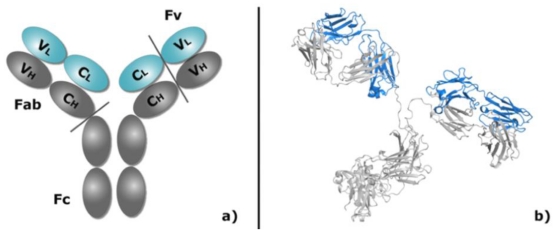
Antibodies play an increasingly important role in both basic research and the pharmaceutical industry. Since their efficiency depends, in ultimate analysis, on their atomic interactions with an antigen, studying such interactions is important to understand how they function and, in the long run, to design new molecules with desired properties. Computational docking, the process of predicting the conformation of a complex from its separated components, is emerging as a fast and affordable technique for the structural characterization of antibody-antigen complexes. In this manuscript, we first describe the different computational strategies for the modeling of antibodies and docking of their complexes, and then predict the binding of two antibodies to the stalk region of influenza hemagglutinin, an important pharmaceutical target. The purpose is two-fold: on a general note, we want to illustrate the advantages and pitfalls of computational docking with a practical example, using different approaches and comparing the results to known experimental structures. On a more specific note, we want to assess if docking can be successful in characterizing the binding to the same influenza epitope of other antibodies with unknown structure, which has practical relevance for pharmaceutical and biological research. The paper clearly shows that some of the computational docking predictions can be very accurate, but the algorithm often fails to discriminate them from inaccurate solutions. It is of paramount importance, therefore, to use rapidly obtained experimental data to validate the computational results.
Keywords: antibody modeling; antibody-antigen complexes; computational docking; hemagglutinin; influenza.
Figures


References
-
- Janin J, Henrick K, Moult J, Eyck LT, Sternberg MJ, Vajda S, Vakser I, Wodak SJ. CAPRI: a Critical Assessment of PRedicted Interactions. Proteins. 2003;52:2–9. - PubMed
-
- Rini JM, Schulze-Gahmen U, Wilson IA. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science. 1992;255:959–965. - PubMed
-
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997;273:927–948. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
