

Apoptotic cell signaling in cancer progression and therapy
- PMID: 21340093
- PMCID: PMC3130501
- DOI: 10.1039/c0ib00144a
Apoptotic cell signaling in cancer progression and therapy
Abstract
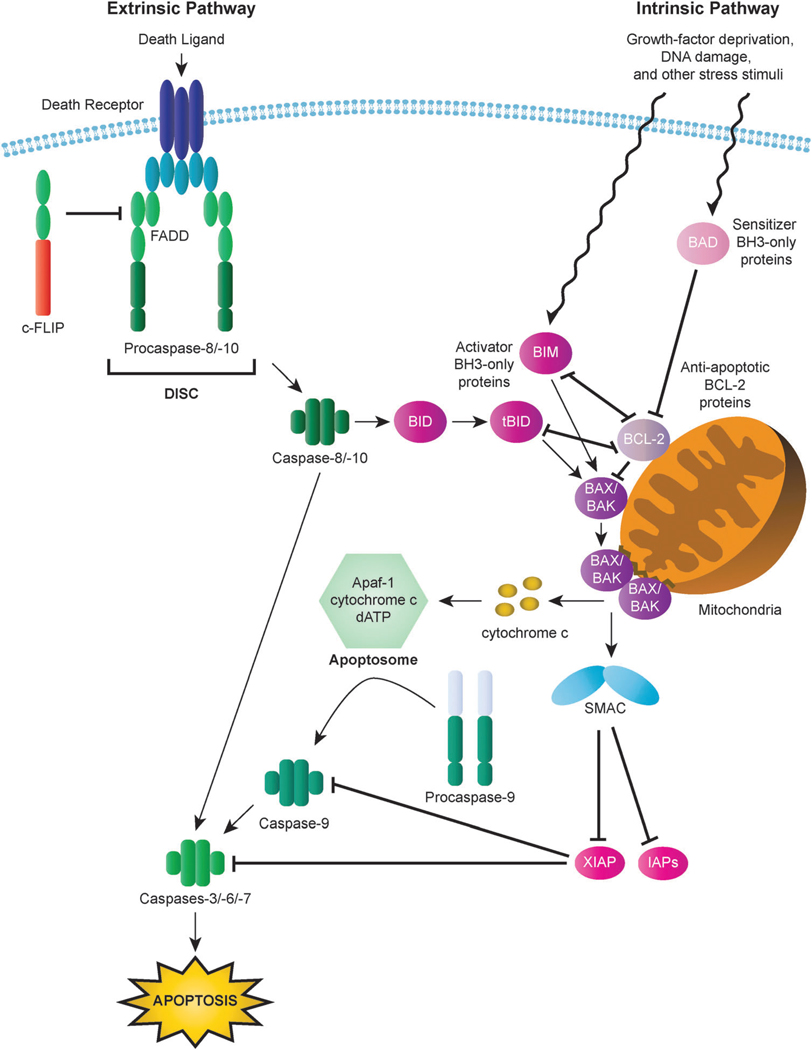
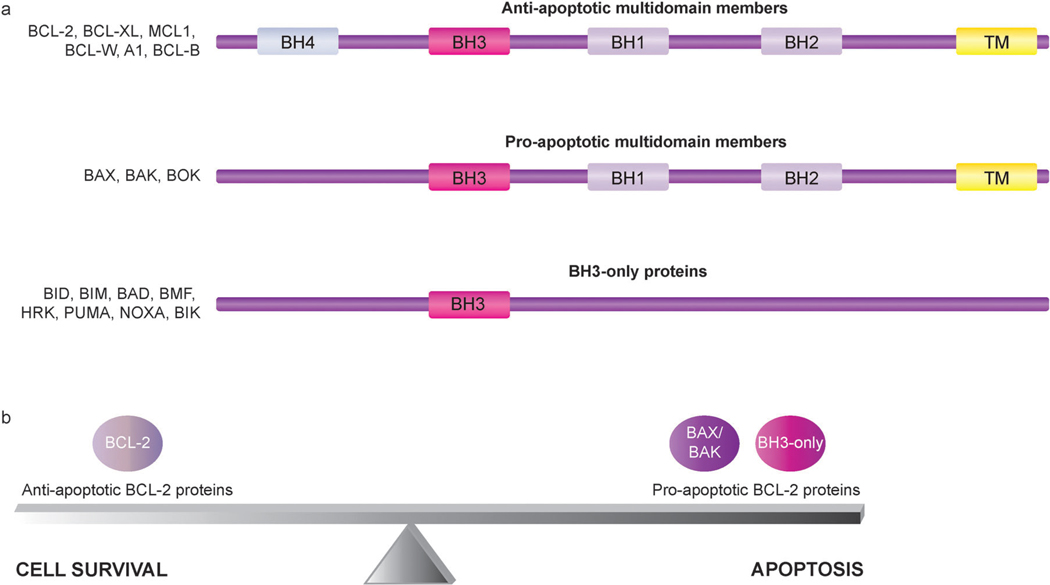
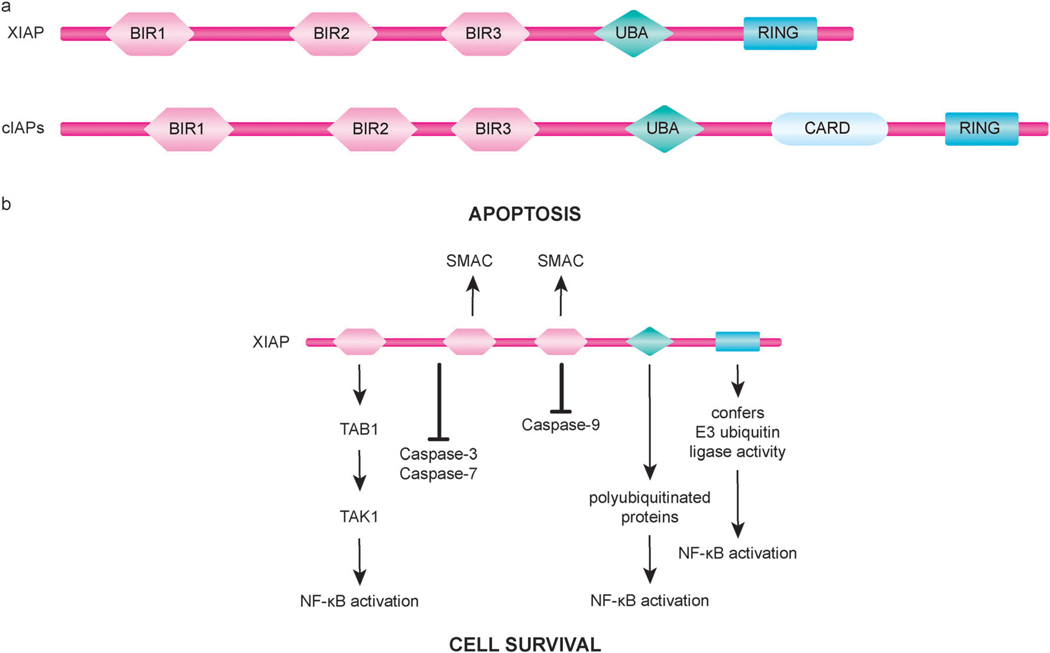
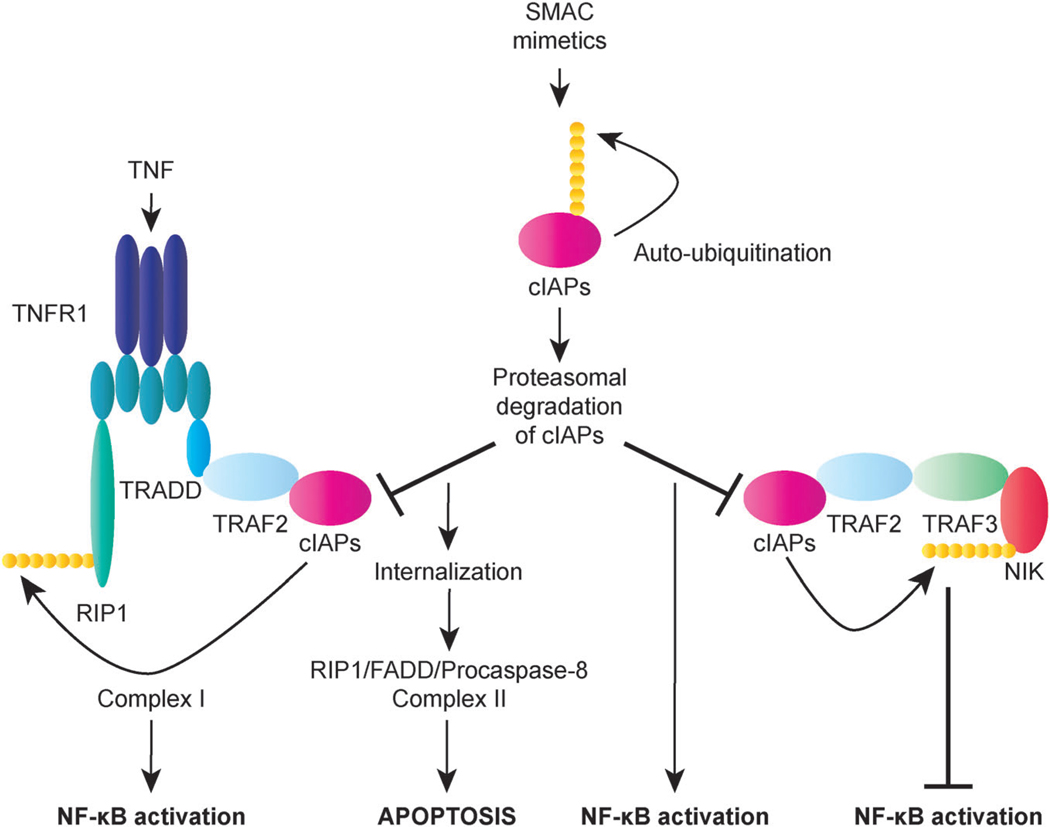
Apoptosis is a tightly regulated cell suicide program that plays an essential role in the development and maintenance of tissue homeostasis by eliminating unnecessary or harmful cells. Impairment of this native defense mechanism promotes aberrant cellular proliferation and the accumulation of genetic defects, ultimately resulting in tumorigenesis, and frequently confers drug resistance to cancer cells. The regulation of apoptosis at several levels is essential to maintain the delicate balance between cellular survival and death signaling that is required to prevent disease. Complex networks of signaling pathways act to promote or inhibit apoptosis in response to various cues. Apoptosis can be triggered by signals from within the cell, such as genotoxic stress, or by extrinsic signals, such as the binding of ligands to cell surface death receptors. Various upstream signaling pathways can modulate apoptosis by converging on, and thereby altering the activity of, common central control points within the apoptotic signaling pathways, which involve the BCL-2 family proteins, inhibitor of apoptosis (IAP) proteins, and FLICE-inhibitory protein (c-FLIP). This review highlights the role of these fundamental regulators of apoptosis in the context of both normal apoptotic signaling mechanisms and dysregulated apoptotic pathways that can render cancer cells resistant to cell death. In addition, therapeutic strategies aimed at modulating the activity of BCL-2 family proteins, IAPs, and c-FLIP for the targeted induction of apoptosis are briefly discussed.
Figures

References
-
- Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat. Rev. Cancer. 2009;9:501–507. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. - PubMed
-
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability-an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
