

A regression analysis of gene expression in ES cells reveals two gene classes that are significantly different in epigenetic patterns
- PMID: 21342583
- PMCID: PMC3044308
- DOI: 10.1186/1471-2105-12-S1-S50
A regression analysis of gene expression in ES cells reveals two gene classes that are significantly different in epigenetic patterns
Abstract
Background: To understand the gene regulatory system that governs the self-renewal and pluripotency of embryonic stem cells (ESCs) is an important step for promoting regenerative medicine. In it, the role of several core transcription factors (TFs), such as Oct4, Sox2 and Nanog, has been intensively investigated, details of their involvement in the genome-wide gene regulation are still not well clarified.
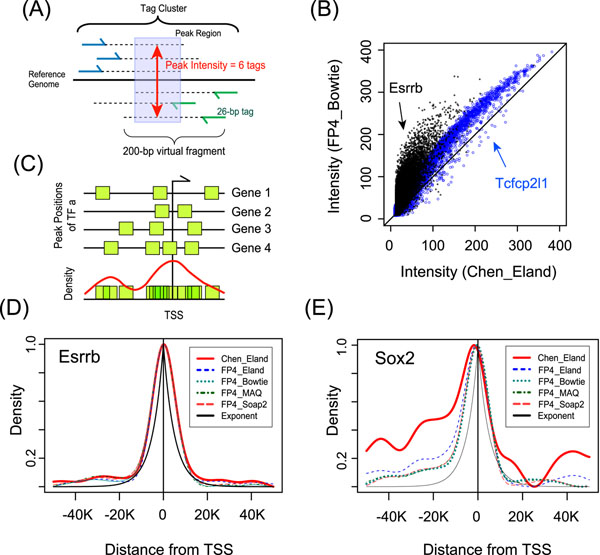
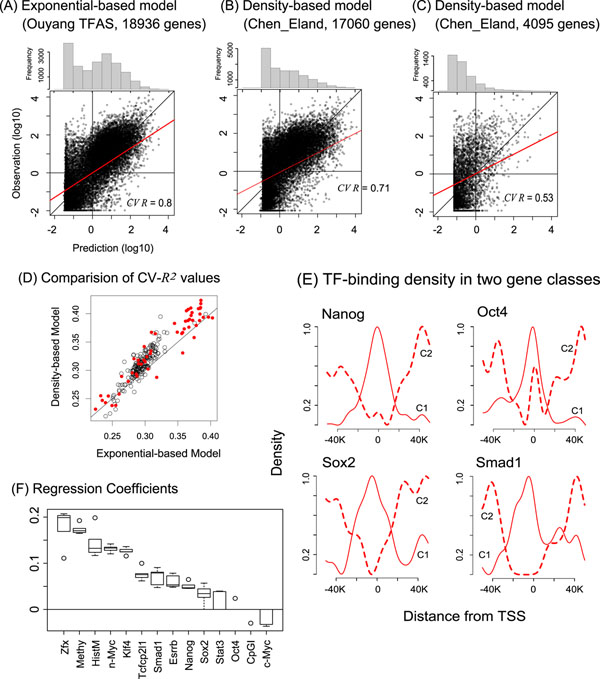
Methods: We constructed a predictive model of genome-wide gene expression in mouse ESCs from publicly available ChIP-seq data of 12 core TFs. The tag sequences were remapped on the genome by various alignment tools. Then, the binding density of each TF is calculated from the genome-wide bona fide TF binding sites. The TF-binding data was combined with the data of several epigenetic states (DNA methylation, several histone modifications, and CpG island) of promoter regions. These data as well as the ordinary peak intensity data were used as predictors of a simple linear regression model that predicts absolute gene expression. We also developed a pipeline for analyzing the effects of predictors and their interactions.
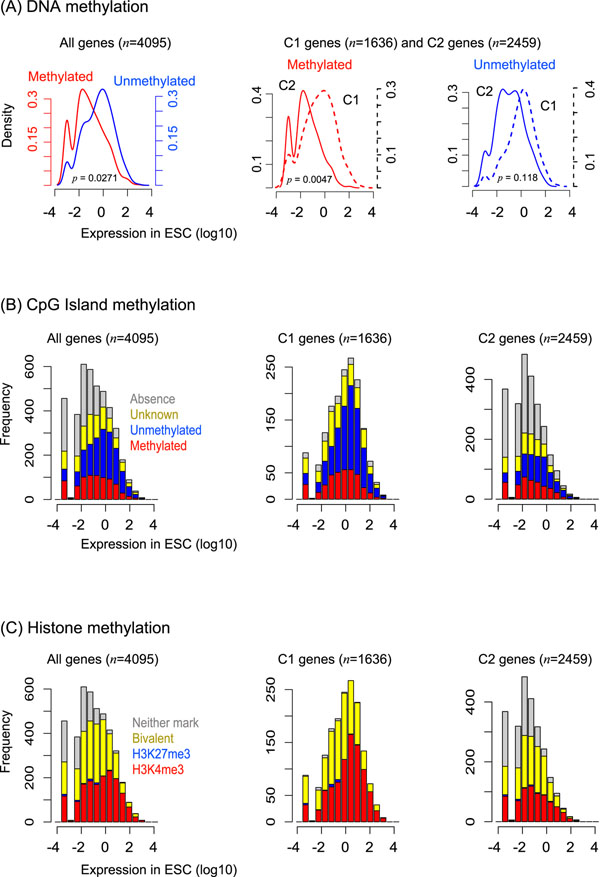
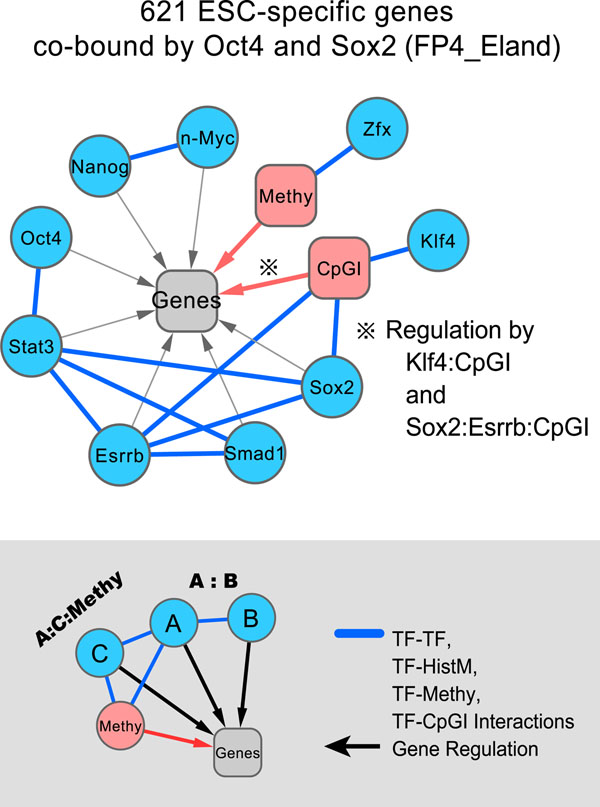
Results: Through our analysis, we identified two classes of genes that are either well explained or inefficiently explained by our model. The latter class seems to be genes that are not directly regulated by the core TFs. The regulatory regions of these gene classes show apparently distinct patterns of DNA methylation, histone modifications, existence of CpG islands, and gene ontology terms, suggesting the relative importance of epigenetic effects. Furthermore, we identified statistically significant TF interactions correlated with the epigenetic modification patterns.
Conclusions: Here, we proposed an improved prediction method in explaining the ESC-specific gene expression. Our study implies that the majority of genes are more or less directly regulated by the core TFs. In addition, our result is consistent with the general idea of relative importance of epigenetic effects in ESCs.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
