

Prevalence and clinical correlates of QT prolongation in patients with hypertrophic cardiomyopathy
- PMID: 21345853
- PMCID: PMC3086898
- DOI: 10.1093/eurheartj/ehr021
Prevalence and clinical correlates of QT prolongation in patients with hypertrophic cardiomyopathy
Abstract
Aims: Congenital or acquired QT prolongation is a risk factor for life-threatening arrhythmias. In patients with hypertrophic cardiomyopathy (HCM), the QT interval may be intrinsically prolonged. However, the prevalence, cause, and significance of QT prolongation among patients with HCM are unknown.
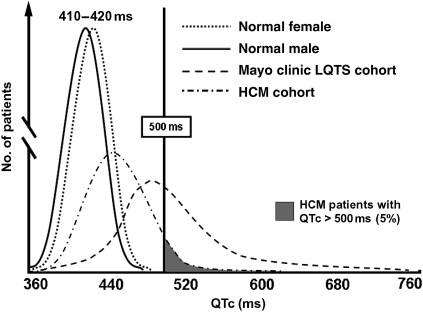
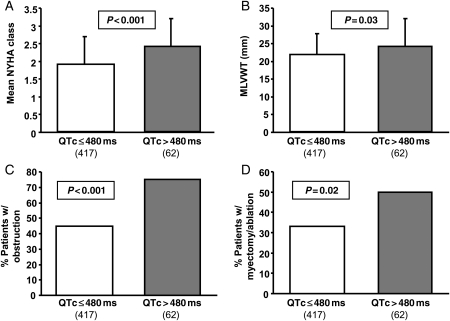
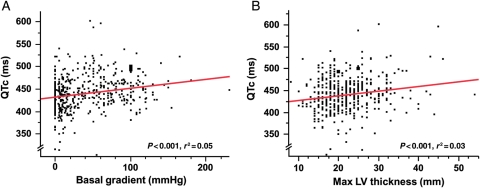
Methods and results: After exclusion of patients on QT-prolonging drugs, a blinded, retrospective analysis of electrocardiograms, echocardiograms, and genotype status in 479 unrelated patients with HCM [201 females, age at diagnosis 41 ± 18 years, maximal left ventricular wall thickness (MLVWT) 22 ± 6 mm] from two independent centres was performed. The mean QTc was 440 ± 28 ms. The QTc exceeded 480 ms in 13% of patients. Age, gender, family history of HCM or sudden cardiac arrest, and genotype status had no association with QTc. Patients with a QTc over 480 ms were more symptomatic at diagnosis (P < 0.001), had a higher MLVWT (P = 0.03), were more obstructive (P < 0.001), and were more likely to have undergone septal reduction therapy (P = 0.02). There was a weak but significant direct linear relationship between QTc and peak outflow gradient (r(2) = 0.05, P < 0.0001).
Conclusions: Compared with <1 in 200 otherwise healthy adults, QT prolongation (QTc > 480 ms) was present in 1 out of 8 patients with HCM. The QTc was partly reflective of the degree of cardiac hypertrophy and left ventricular outflow tract obstruction. Because of its pro-arrhythmic potential and its potential relevance to management and risk stratification, routine QTc assessment should be performed in patients with HCM, particularly when concomitant use of QT-prolonging medications is considered.
Figures
References
-
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi:10.1001/jama.287.10.1308. - DOI - PubMed
-
- Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. doi:10.1016/j.jacc.2009.02.075. - DOI - PubMed
-
- Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, Epstein AE, Almquist AK, Daubert JP, Lawrenz T, Boriani G, Estes NA, 3rd, Favale S, Piccininno M, Winters SL, Santini M, Betocchi S, Arribas F, Sherrid MV, Buja G, Semsarian C, Bruzzi P. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405–412. doi:10.1001/jama.298.4.405. - DOI - PubMed
-
- McKenna WJ, Goodwin JF. The natural history of hypertrophic cardiomyopathy. Curr Probl Cardiol. 1981;6:1–26. doi:10.1016/0146-2806(81)90015-3. - DOI - PubMed
-
- Spirito P, Autore C, Rapezzi C, Bernabo P, Badagliacca R, Maron MS, Bongioanni S, Coccolo F, Estes NA, Barilla CS, Biagini E, Quarta G, Conte MR, Bruzzi P, Maron BJ. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703–1710. doi:10.1161/CIRCULATIONAHA.108.798314. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
