

Epistatic interaction maps relative to multiple metabolic phenotypes
- PMID: 21347328
- PMCID: PMC3037399
- DOI: 10.1371/journal.pgen.1001294
Epistatic interaction maps relative to multiple metabolic phenotypes
Abstract
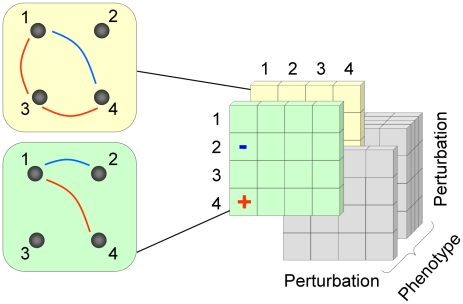
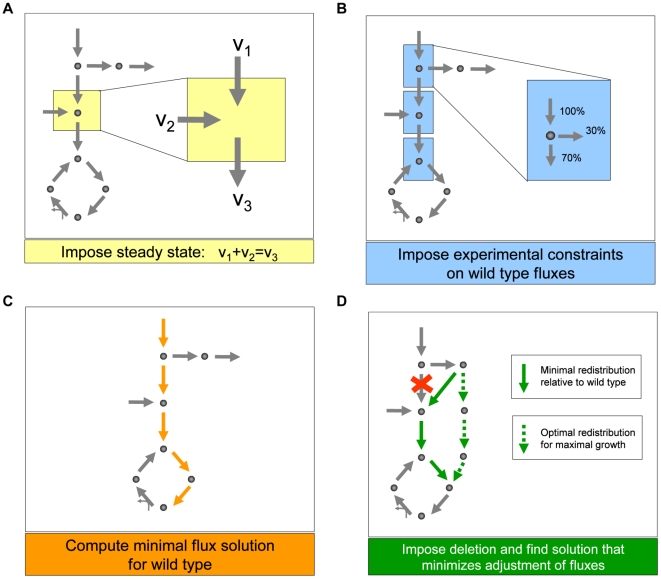
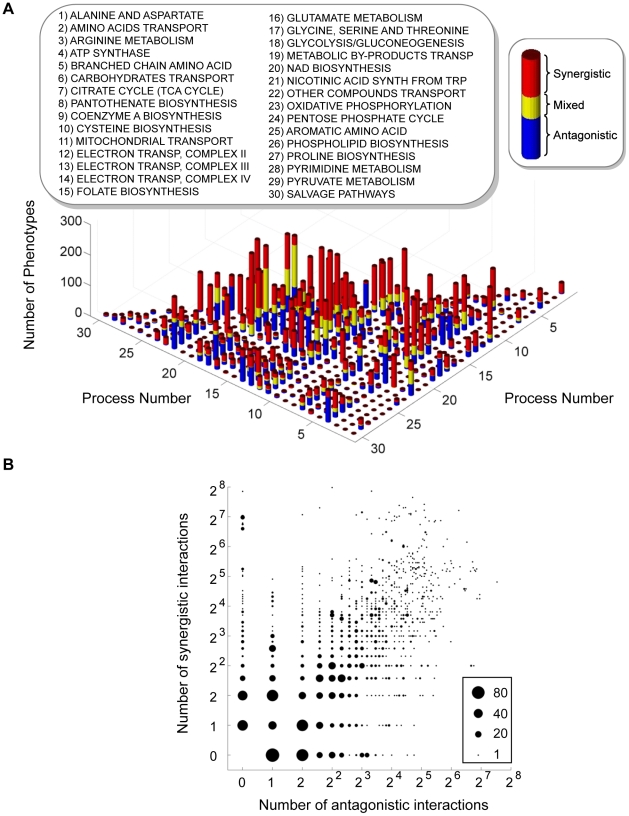
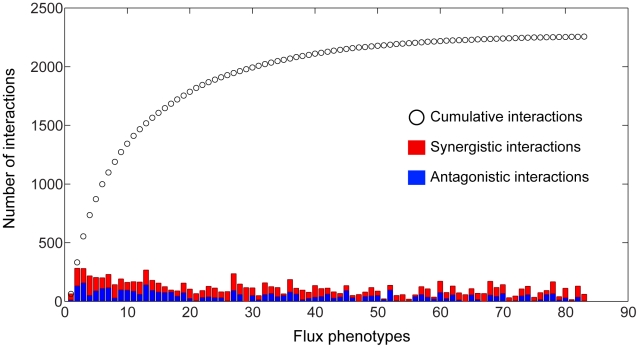
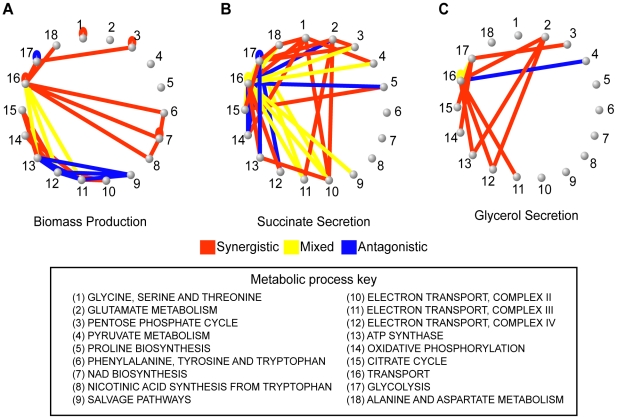
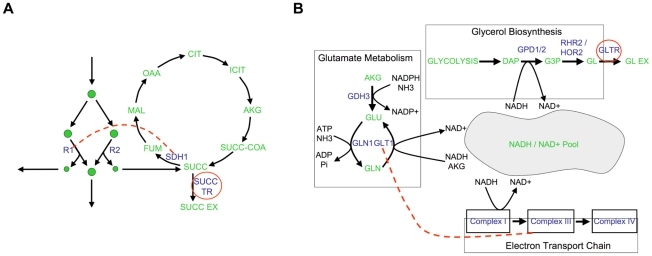
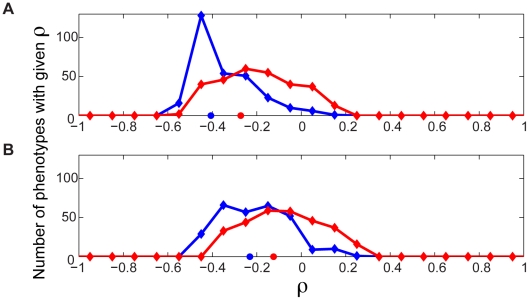
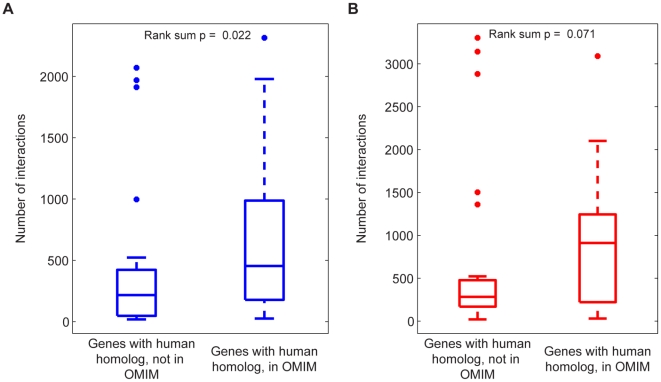
An epistatic interaction between two genes occurs when the phenotypic impact of one gene depends on another gene, often exposing a functional association between them. Due to experimental scalability and to evolutionary significance, abundant work has been focused on studying how epistasis affects cellular growth rate, most notably in yeast. However, epistasis likely influences many different phenotypes, affecting our capacity to understand cellular functions, biochemical networks adaptation, and genetic diseases. Despite its broad significance, the extent and nature of epistasis relative to different phenotypes remain fundamentally unexplored. Here we use genome-scale metabolic network modeling to investigate the extent and properties of epistatic interactions relative to multiple phenotypes. Specifically, using an experimentally refined stoichiometric model for Saccharomyces cerevisiae, we computed a three-dimensional matrix of epistatic interactions between any two enzyme gene deletions, with respect to all metabolic flux phenotypes. We found that the total number of epistatic interactions between enzymes increases rapidly as phenotypes are added, plateauing at approximately 80 phenotypes, to an overall connectivity that is roughly 8-fold larger than the one observed relative to growth alone. Looking at interactions across all phenotypes, we found that gene pairs interact incoherently relative to different phenotypes, i.e. antagonistically relative to some phenotypes and synergistically relative to others. Specific deletion-deletion-phenotype triplets can be explained metabolically, suggesting a highly informative role of multi-phenotype epistasis in mapping cellular functions. Finally, we found that genes involved in many interactions across multiple phenotypes are more highly expressed, evolve slower, and tend to be associated with diseases, indicating that the importance of genes is hidden in their total phenotypic impact. Our predictions indicate a pervasiveness of nonlinear effects in how genetic perturbations affect multiple metabolic phenotypes. The approaches and results reported could influence future efforts in understanding metabolic diseases and the role of biochemical regulation in the cell.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Modular epistasis and the compensatory evolution of gene deletion mutants.PLoS Genet. 2019 Feb 15;15(2):e1007958. doi: 10.1371/journal.pgen.1007958. eCollection 2019 Feb. PLoS Genet. 2019. PMID: 30768593 Free PMC article.
-
Prevalent positive epistasis in Escherichia coli and Saccharomyces cerevisiae metabolic networks.Nat Genet. 2010 Mar;42(3):272-6. doi: 10.1038/ng.524. Epub 2010 Jan 24. Nat Genet. 2010. PMID: 20101242 Free PMC article.
-
Dynamic epistasis under varying environmental perturbations.PLoS One. 2015 Jan 27;10(1):e0114911. doi: 10.1371/journal.pone.0114911. eCollection 2015. PLoS One. 2015. PMID: 25625594 Free PMC article.
-
Organization principles in genetic interaction networks.Adv Exp Med Biol. 2012;751:53-78. doi: 10.1007/978-1-4614-3567-9_3. Adv Exp Med Biol. 2012. PMID: 22821453 Review.
-
Exploring genetic interactions and networks with yeast.Nat Rev Genet. 2007 Jun;8(6):437-49. doi: 10.1038/nrg2085. Nat Rev Genet. 2007. PMID: 17510664 Review.
Cited by
-
Have you tried turning it off and on again? Oscillating selection to enhance fitness-landscape traversal in adaptive laboratory evolution experiments.Metab Eng Commun. 2023 Jul 13;17:e00227. doi: 10.1016/j.mec.2023.e00227. eCollection 2023 Dec. Metab Eng Commun. 2023. PMID: 37538933 Free PMC article. Review.
-
Impact of stoichiometry representation on simulation of genotype-phenotype relationships in metabolic networks.PLoS Comput Biol. 2012;8(11):e1002758. doi: 10.1371/journal.pcbi.1002758. Epub 2012 Nov 1. PLoS Comput Biol. 2012. PMID: 23133362 Free PMC article.
-
Computational design and engineering of an Escherichia coli strain producing the nonstandard amino acid para-aminophenylalanine.iScience. 2022 Jun 9;25(7):104562. doi: 10.1016/j.isci.2022.104562. eCollection 2022 Jul 15. iScience. 2022. PMID: 35789833 Free PMC article.
-
RELATCH: relative optimality in metabolic networks explains robust metabolic and regulatory responses to perturbations.Genome Biol. 2012 Jul 5;13(9):R78. doi: 10.1186/gb-2012-13-9-r78. Genome Biol. 2012. PMID: 23013597 Free PMC article.
-
What mRNA Abundances Can Tell us about Metabolism.Metabolites. 2012 Sep 12;2(3):614-31. doi: 10.3390/metabo2030614. Metabolites. 2012. PMID: 24957650 Free PMC article.
References
-
- Guarente L. Synthetic enhancement in gene interaction: a genetic tool come of age. Trends Genet. 1993;9:362–366. - PubMed
-
- Stein A, Aloy P. A molecular interpretation of genetic interactions in yeast. FEBS Lett. In Press, Uncorrected Proof. Available at: http://www.sciencedirect.com/science/article/B6T36-4RW9H0W-9/2/65bd6932e.... Accessed 25 February 2008. - PubMed
-
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, et al. Exploration of the Function and Organization of the Yeast Early Secretory Pathway through an Epistatic Miniarray Profile. Cell. 2005;123:507–519. - PubMed
-
- Ye P, Peyser BD, Pan X, Boeke JD, Spencer FA, et al. Gene function prediction from congruent synthetic lethal interactions in yeast. Mol Syst Biol 1 . 2005. Available at: http://dx.doi.org/10.1038/msb4100034. Accessed 4 December 2008. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
