

Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy
- PMID: 21353195
- PMCID: PMC3059419
- DOI: 10.1016/j.ajhg.2011.01.016
Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy
Abstract
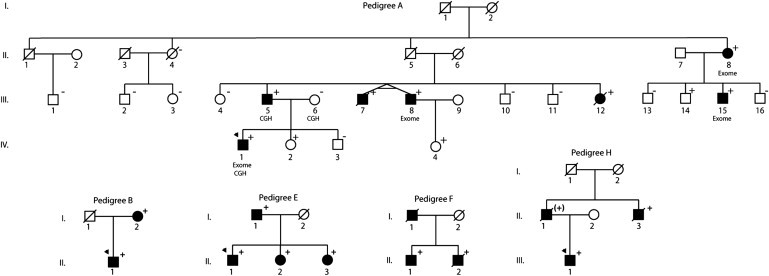
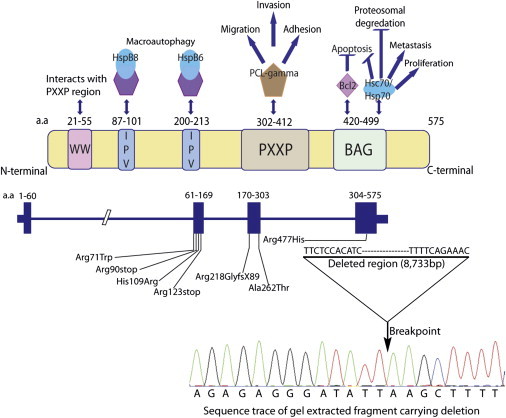
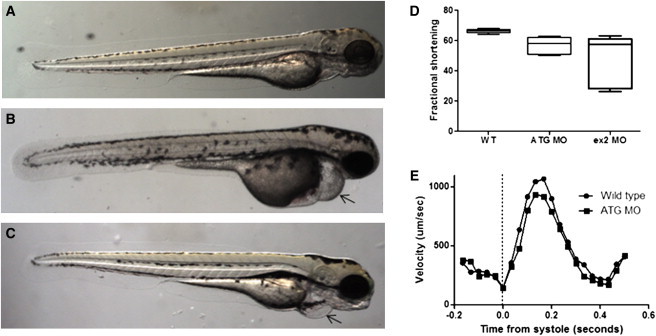
Dilated cardiomyopathy commonly causes heart failure and is the most frequent precipitating cause of heart transplantation. Familial dilated cardiomyopathy has been shown to be caused by rare variant mutations in more than 30 genes but only ~35% of its genetic cause has been identified, principally by using linkage-based or candidate gene discovery approaches. In a multigenerational family with autosomal dominant transmission, we employed whole-exome sequencing in a proband and three of his affected family members, and genome-wide copy number variation in the proband and his affected father and unaffected mother. Exome sequencing identified 428 single point variants resulting in missense, nonsense, or splice site changes. Genome-wide copy number analysis identified 51 insertion deletions and 440 copy number variants > 1 kb. Of these, a 8733 bp deletion, encompassing exon 4 of the heat shock protein cochaperone BCL2-associated athanogene 3 (BAG3), was found in seven affected family members and was absent in 355 controls. To establish the relevance of variants in this protein class in genetic DCM, we sequenced the coding exons in BAG3 in 311 other unrelated DCM probands and identified one frameshift, two nonsense, and four missense rare variants absent in 355 control DNAs, four of which were familial and segregated with disease. Knockdown of bag3 in a zebrafish model recapitulated DCM and heart failure. We conclude that new comprehensive genomic approaches have identified rare variants in BAG3 as causative of DCM.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Hershberger R.E., Parks S.B., Kushner J.D., Li D., Ludwigsen S., Jakobs P., Nauman D., Burgess D., Partain J., Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–26. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
