

Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta
- PMID: 21353196
- PMCID: PMC3059418
- DOI: 10.1016/j.ajhg.2011.01.015
Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta
Abstract
Osteogenesis imperfecta (OI) is a heterogeneous genetic disorder characterized by bone fragility and susceptibility to fractures after minimal trauma. After mutations in all known OI genes had been excluded by Sanger sequencing, we applied next-generation sequencing to analyze the exome of a single individual who has a severe form of the disease and whose parents are second cousins. A total of 26,922 variations from the human reference genome sequence were subjected to several filtering steps. In addition, we extracted the genotypes of all dbSNP130-annotated SNPs from the exome sequencing data and used these 299,494 genotypes as markers for the genome-wide identification of homozygous regions. A single homozygous truncating mutation, affecting SERPINF1 on chromosome 17p13.3, that was embedded into a homozygous stretch of 2.99 Mb remained. The mutation was also homozygous in the affected brother of the index patient. Subsequently, we identified homozygosity for two different truncating SERPINF1 mutations in two unrelated patients with OI and parental consanguinity. All four individuals with SERPINF1 mutations have severe OI. Fractures of long bones and severe vertebral compression fractures with resulting deformities were observed as early as the first year of life in these individuals. Collagen analyses with cultured dermal fibroblasts displayed no evidence for impaired collagen folding, posttranslational modification, or secretion. SERPINF1 encodes pigment epithelium-derived factor (PEDF), a secreted glycoprotein of the serpin superfamily. PEDF is a multifunctional protein and one of the strongest inhibitors of angiogenesis currently known in humans. Our data provide genetic evidence for PEDF involvement in human bone homeostasis.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
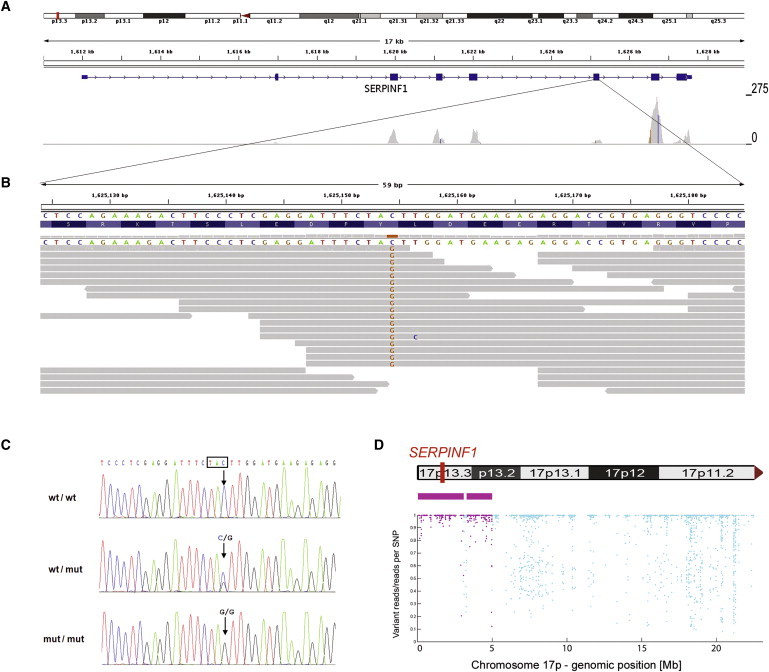
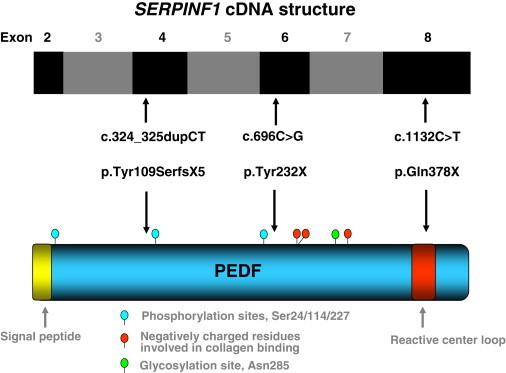

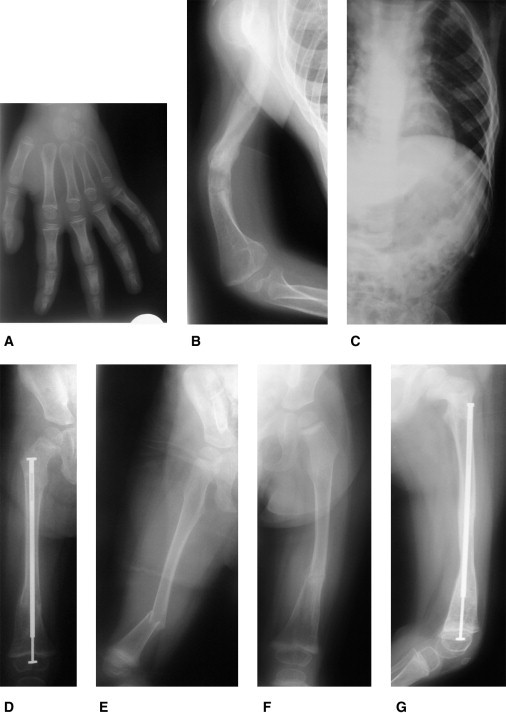
References
-
- Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. - PubMed
-
- Basel D., Steiner R.D. Osteogenesis imperfecta: Recent findings shed new light on this once well-understood condition. Genet. Med. 2009;11:375–385. - PubMed
-
- Chu M.L., Williams C.J., Pepe G., Hirsch J.L., Prockop D.J., Ramirez F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304:78–80. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
