

Review
doi: 10.1016/j.sbi.2011.02.001.
Epub 2011 Feb 24.
Protein function prediction: towards integration of similarity metrics
Affiliations
- PMID: 21353529
- PMCID: PMC3120633
- DOI: 10.1016/j.sbi.2011.02.001
Item in Clipboard
Review
Protein function prediction: towards integration of similarity metrics
Curr Opin Struct Biol.
2011 Apr.
Abstract
Genomic centers discover increasingly many protein sequences and structures, but not necessarily their full biological functions. Thus, currently, less than one percent of proteins have experimentally verified biochemical activities. To fill this gap, function prediction algorithms apply metrics of similarity between proteins on the premise that those sufficiently alike in sequence, or structure, will perform identical functions. Although high sensitivity is elusive, network analyses that integrate these metrics together hold the promise of rapid gains in function prediction specificity.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
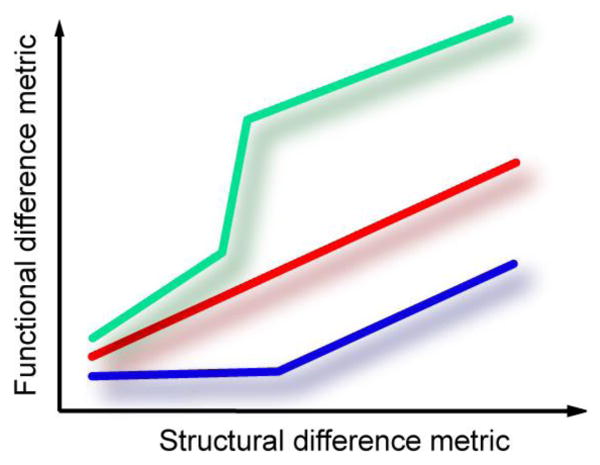
The x-axis represents the distance between two proteins, here in term of structure—but a metric based on sequence or on some other observable feature would have similar features. The y-axis is the distance between the same proteins in terms of their biological functions. Typically, annotations methods assume that the more similar the proteins the more alike their function. This is shown as a simple (linear) correlation in the red line. But these changes need not to be smooth: the green line illustrates small protein variations that lead to substantial change in molecular function, such as between paralogs. The blue line illustrates an opposite example when distant proteins perform closely related biochemical functions.

A. ETA is composed of three steps. 1) The Evolutionary Trace [55] aligns homologous sequences and ranks positions according to the correlation between evolutionary divergence and amino acid variations. 2) The protein structure is labeled with these evolutionary importance rankings. 3) A heuristic selects clustered, surface exposed and evolutionarily important amino acids to form a structural template (red spheres). 4) A library of proteins with known function is searched for matches (called hits) to this template. An SVM filters discards the hits if they do not fall on top ranked ET residues (not depicted). 5-8) A reciprocal match is searched for and here shown to be found by repeating steps 1-4 in the opposite direction. B. ETA matches define a graph. Each protein chains is a node, and structural and evolutionary similarities are the edges. Some nodes are known to carry a given function (blue), other nodes are known to not carry that function (white), and the functional status of remaining nodes is unknown (?). The labels are then transferred among all nodes in the network based on the number of edges and their strength, in a process analogous to diffusion. The result is a score for every enzymatic function at every node. Finally, these scores are normalized and compared (not depicted). The predicted functional label is the one with the highest normalized weight (called z-score) that is also significant. C. Performance comparison of ETA network diffusion versus BLAST on a test set of structural genomics proteins. Diffusion of enzymatic function annotations showed a consistent accuracy advantage of approximately 9% over BLAST across many coverage levels [80]. D. UV absorbance (y-axis) confirms the predicted carboxylesterase activity of a previously unannotated protein from the medically relevant organism Staphylococcus aureus (3h04 in the Protein Data Bank). ETA network diffusion predicted this enzymatic function which was tested and confirmed in vitro. Specific activity was similar to that of a known carboxylesterase; the negative control, Bovine serum albumin (BSA), had no activity.
References
-
- Rost B. Enzyme function less conserved than anticipated. J Mol Biol. 2002;318:595–608. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
