

Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice
- PMID: 21355090
- PMCID: PMC3100702
- DOI: 10.1182/blood-2010-11-316885
Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice
Abstract
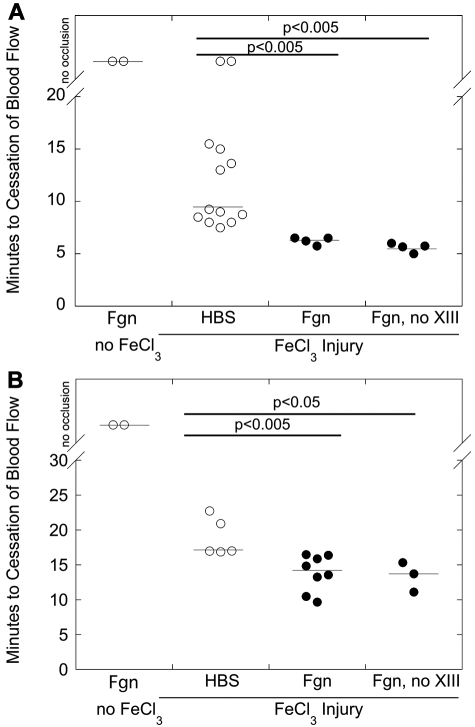
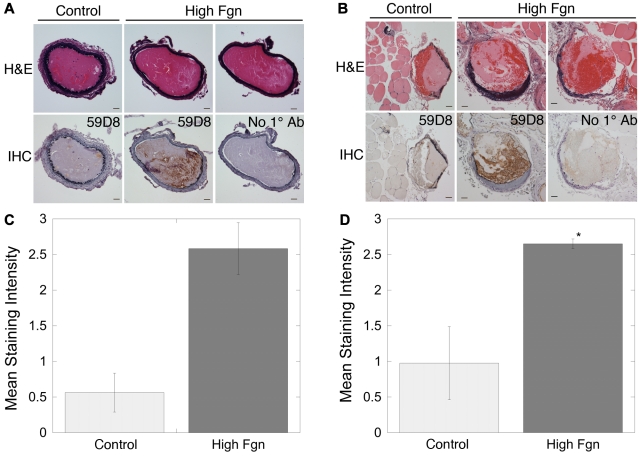
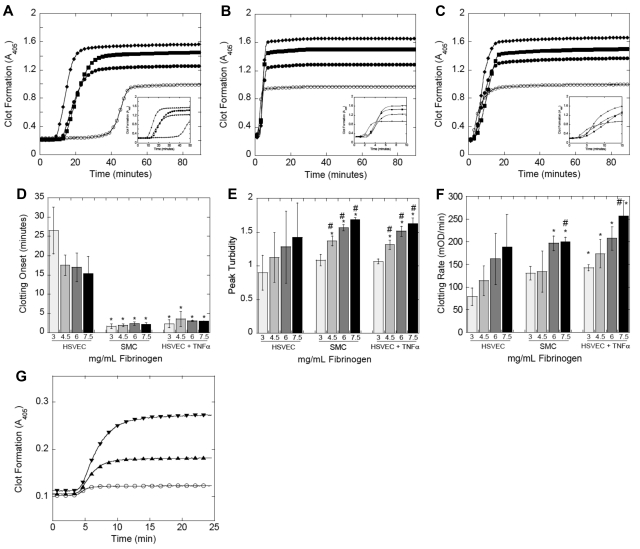
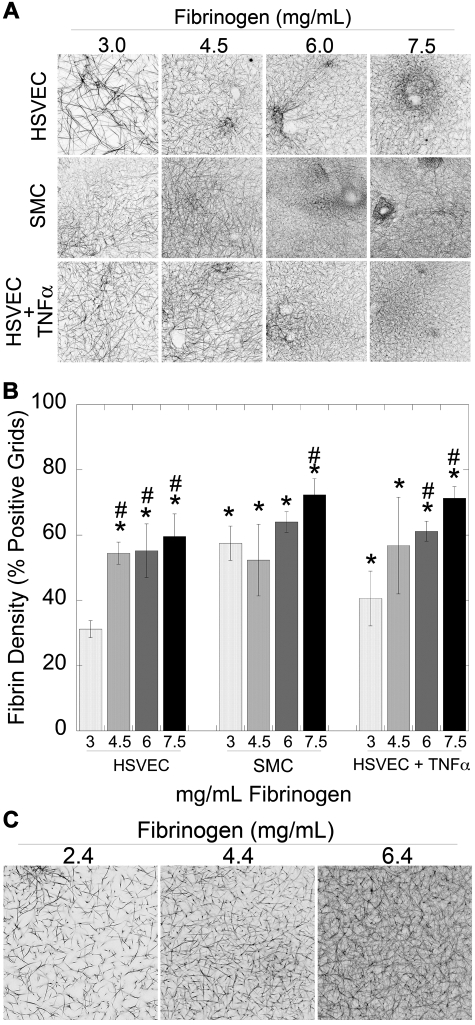
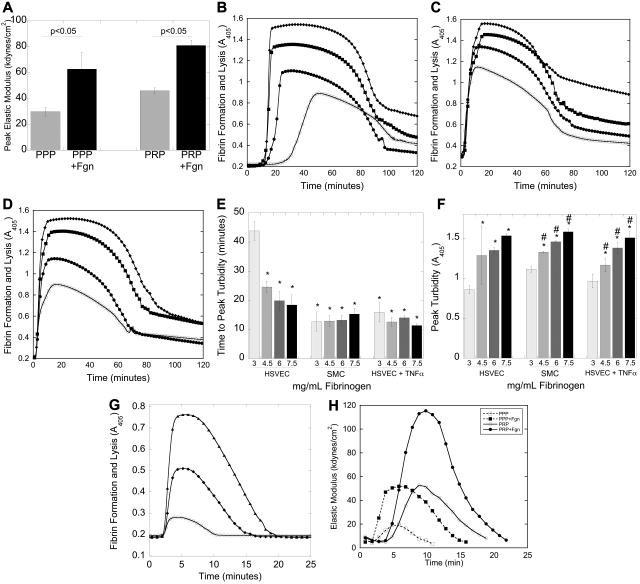
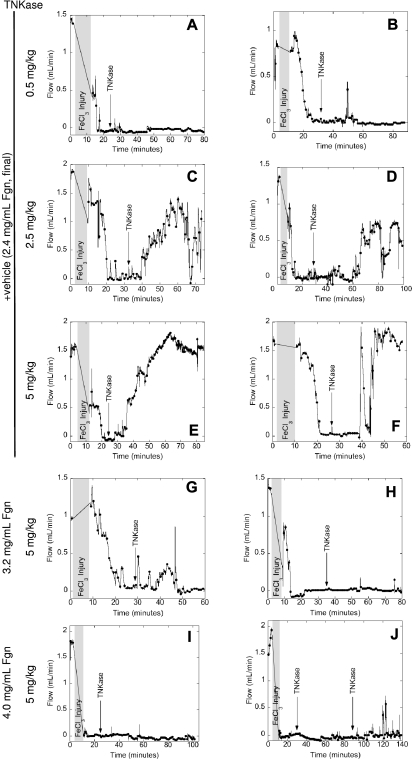
Epidemiologic studies have correlated elevated plasma fibrinogen (hyperfibrinogenemia) with risk of cardiovascular disease and arterial and venous thrombosis. However, it is unknown whether hyperfibrinogenemia is merely a biomarker of the proinflammatory disease state or is a causative mechanism in the etiology. We raised plasma fibrinogen levels in mice via intravenous infusion and induced thrombosis by ferric chloride application to the carotid artery (high shear) or saphenous vein (lower shear); hyperfibrinogenemia significantly shortened the time to occlusion in both models. Using immunohistochemistry, turbidity, confocal microscopy, and elastometry of clots produced in cell and tissue factor-initiated models of thrombosis, we show that hyperfibrinogenemia increased thrombus fibrin content, promoted faster fibrin formation, and increased fibrin network density, strength, and stability. Hyperfibrinogenemia also increased thrombus resistance to tenecteplase-induced thrombolysis in vivo. These data indicate that hyperfibrinogenemia directly promotes thrombosis and thrombolysis resistance and does so via enhanced fibrin formation and stability. These findings strongly suggest a causative role for hyperfibrinogenemia in acute thrombosis and have significant implications for thrombolytic therapy. Plasma fibrinogen levels may be used to identify patients at risk for thrombosis and inform thrombolytic administration for treating acute thrombosis/thromboembolism.
Figures
Comment in
-
Elevated fibrinogen causes thrombosis.Blood. 2011 May 5;117(18):4687-8. doi: 10.1182/blood-2011-03-340422. Blood. 2011. PMID: 21546470 No abstract available.
References
-
- Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease: FRISC Study Group. N Engl J Med. 2000;343(16):1139–1147. - PubMed
-
- Danesh J, Lewington S, Thompson SG, et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294(14):1799–1809. - PubMed
-
- Wilhelmsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311(8):501–505. - PubMed
-
- Acevedo M, Pearce GL, Kottke-Marchant K, Sprecher DL. Elevated fibrinogen and homocysteine levels enhance the risk of mortality in patients from a high-risk preventive cardiology clinic. Arterioscler Thromb Vasc Biol. 2002;22(6):1042–1045. - PubMed
-
- van Hylckama Vlieg A, Rosendaal FR. High levels of fibrinogen are associated with the risk of deep venous thrombosis mainly in the elderly. J Thromb Haemost. 2003;1(12):2677–2678. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
