

Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A
- PMID: 21355092
- PMCID: PMC3112046
- DOI: 10.1182/blood-2010-10-311589
Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A
Abstract
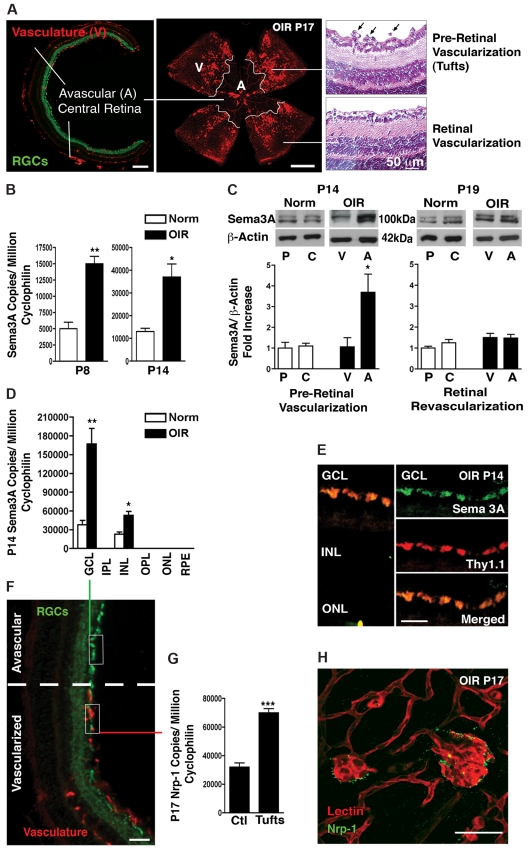
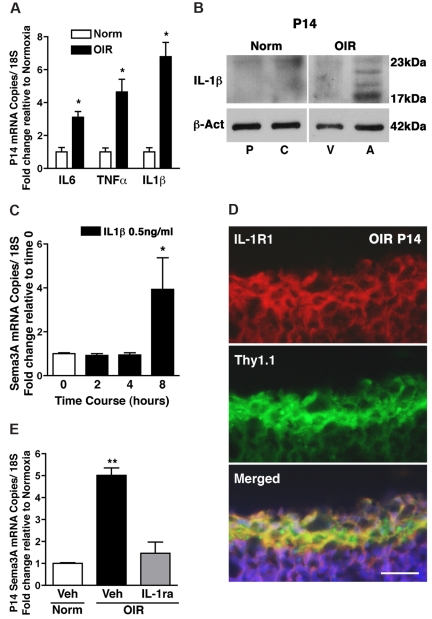
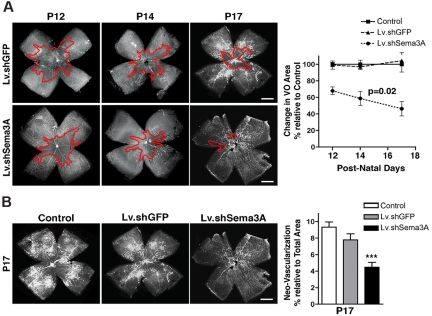
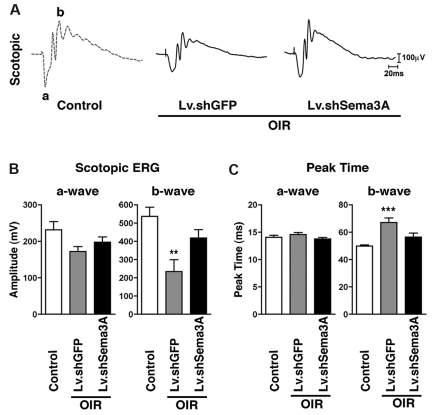
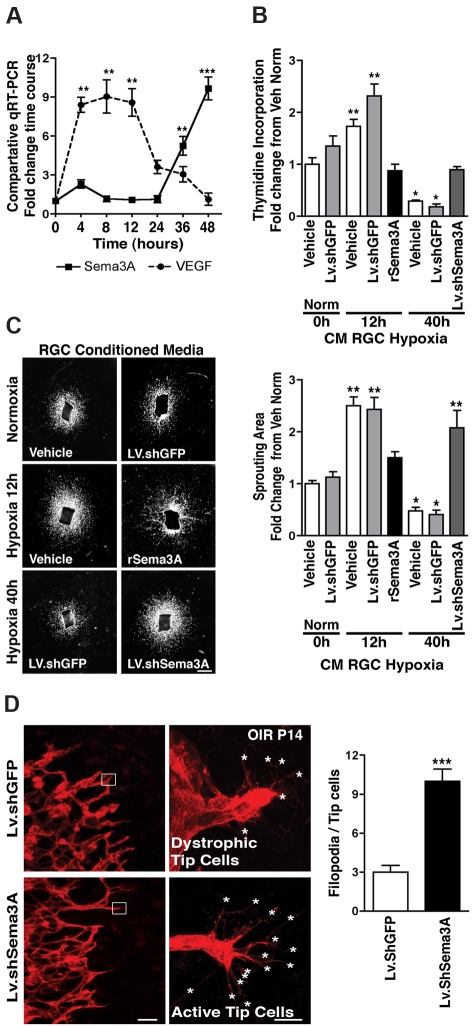
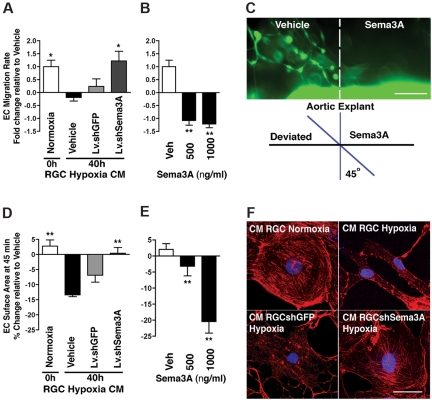
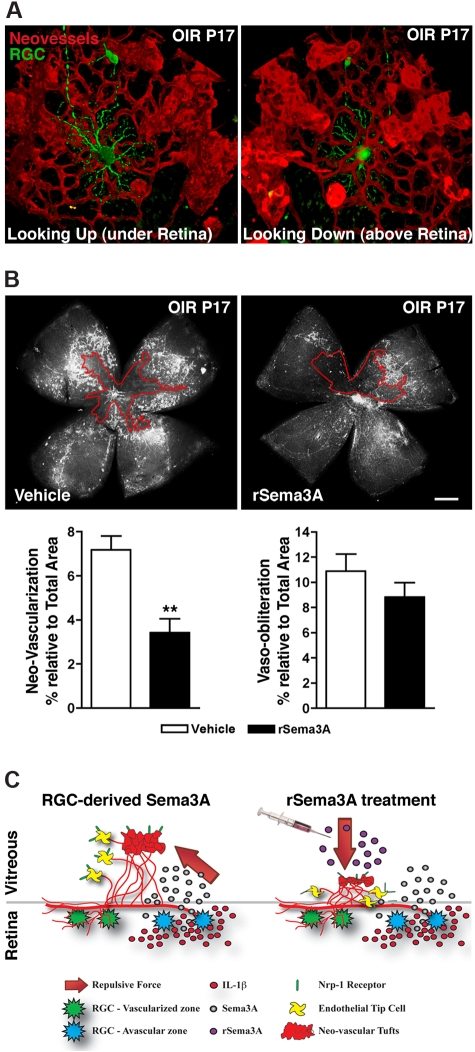
The failure of blood vessels to revascularize ischemic neural tissue represents a significant challenge for vascular biology. Examples include proliferative retinopathies (PRs) such as retinopathy of prematurity and proliferative diabetic retinopathy, which are the leading causes of blindness in children and working-age adults. PRs are characterized by initial microvascular degeneration, followed by a compensatory albeit pathologic hypervascularization mounted by the hypoxic retina attempting to reinstate metabolic equilibrium. Paradoxically, this secondary revascularization fails to grow into the most ischemic regions of the retina. Instead, the new vessels are misdirected toward the vitreous, suggesting that vasorepulsive forces operate in the avascular hypoxic retina. In the present study, we demonstrate that the neuronal guidance cue semaphorin 3A (Sema3A) is secreted by hypoxic neurons in the avascular retina in response to the proinflammatory cytokine IL-1β. Sema3A contributes to vascular decay and later forms a chemical barrier that repels neo-vessels toward the vitreous. Conversely, silencing Sema3A expression enhances normal vascular regeneration within the ischemic retina, thereby diminishing aberrant neovascularization and preserving neuroretinal function. Overcoming the chemical barrier (Sema3A) released by ischemic neurons accelerates the vascular regeneration of neural tissues, which restores metabolic supply and improves retinal function. Our findings may be applicable to other neurovascular ischemic conditions such as stroke.
Figures
Comment in
-
Sema3A [corrected] resists retinal revascularization.Blood. 2011 Jun 2;117(22):5785-6. doi: 10.1182/blood-2011-03-343228. Blood. 2011. PMID: 21636713
References
-
- Sapieha P, Sirinyan M, Hamel D, et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med. 2008;14(10):1067–1076. - PubMed
-
- Dorfman A, Dembinska O, Chemtob S, Lachapelle P. Early manifestations of postnatal hyperoxia on the retinal structure and function of the neonatal rat. Invest Ophthalmol Vis Sci. 2008;49(1):458–466. - PubMed
-
- Dorfman AL, Polosa A, Joly S, Chemtob S, Lachapelle P. Functional and structural changes resulting from strain differences in the rat model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2009;50(5):2436–2450. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
