

Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds
- PMID: 21356090
- PMCID: PMC3056800
- DOI: 10.1186/1471-2164-12-131
Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds
Abstract
Background: Tea is one of the most popular non-alcoholic beverages worldwide. However, the tea plant, Camellia sinensis, is difficult to culture in vitro, to transform, and has a large genome, rendering little genomic information available. Recent advances in large-scale RNA sequencing (RNA-seq) provide a fast, cost-effective, and reliable approach to generate large expression datasets for functional genomic analysis, which is especially suitable for non-model species with un-sequenced genomes.
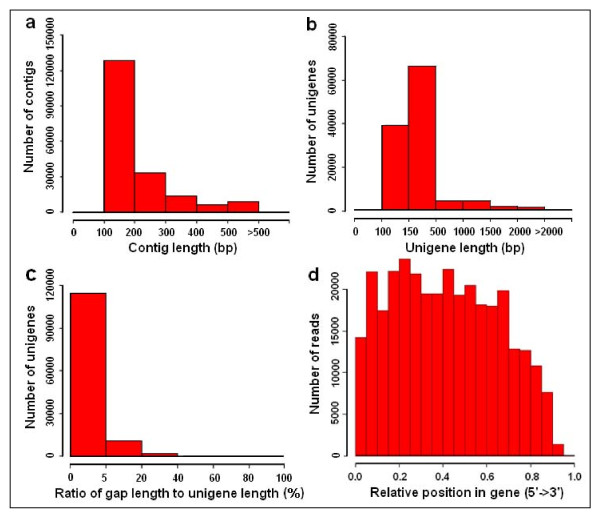
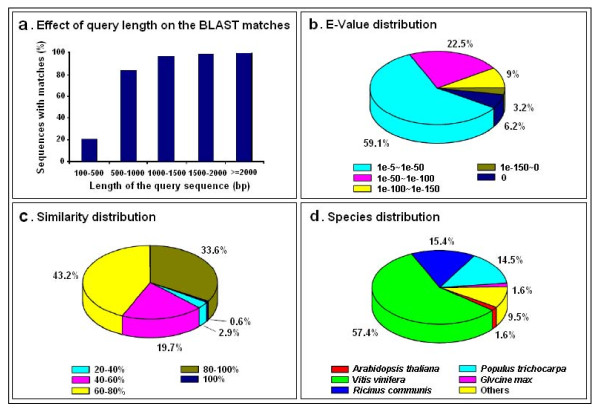
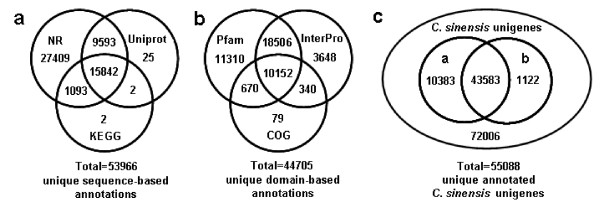
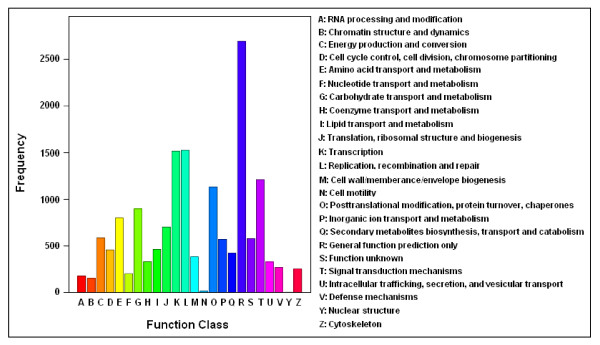
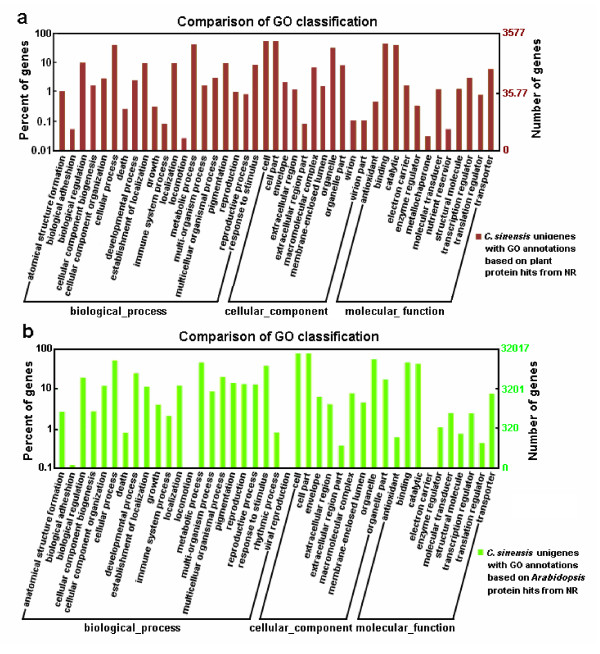
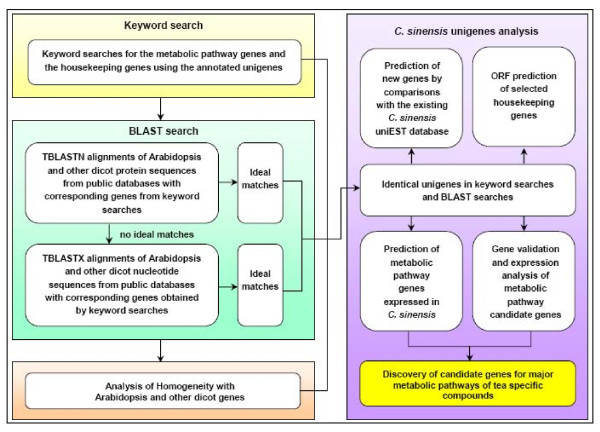
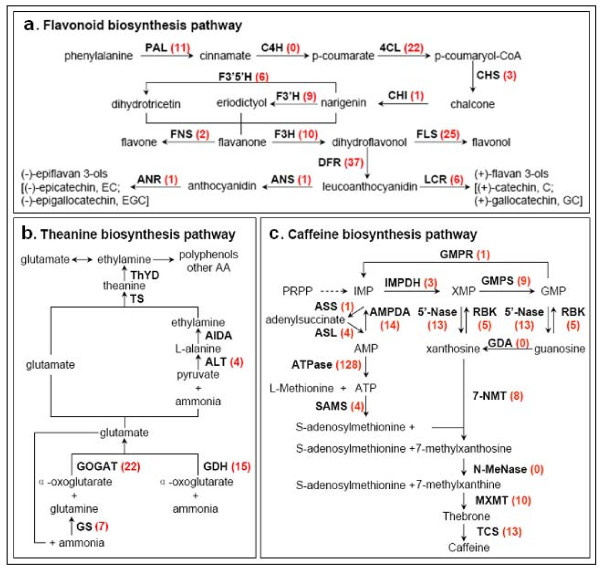
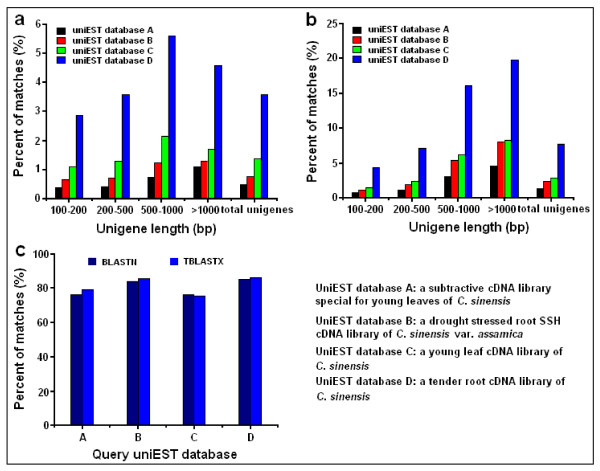
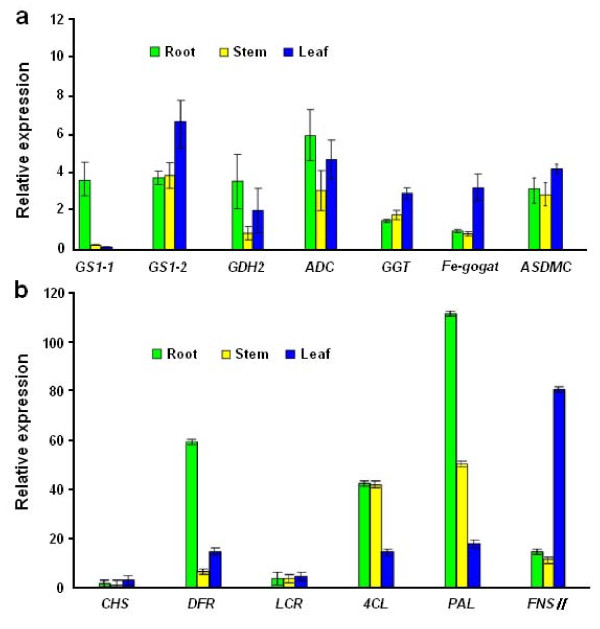
Results: Using high-throughput Illumina RNA-seq, the transcriptome from poly (A)+ RNA of C. sinensis was analyzed at an unprecedented depth (2.59 gigabase pairs). Approximate 34.5 million reads were obtained, trimmed, and assembled into 127,094 unigenes, with an average length of 355 bp and an N50 of 506 bp, which consisted of 788 contig clusters and 126,306 singletons. This number of unigenes was 10-fold higher than existing C. sinensis sequences deposited in GenBank (as of August 2010). Sequence similarity analyses against six public databases (Uniprot, NR and COGs at NCBI, Pfam, InterPro and KEGG) found 55,088 unigenes that could be annotated with gene descriptions, conserved protein domains, or gene ontology terms. Some of the unigenes were assigned to putative metabolic pathways. Targeted searches using these annotations identified the majority of genes associated with several primary metabolic pathways and natural product pathways that are important to tea quality, such as flavonoid, theanine and caffeine biosynthesis pathways. Novel candidate genes of these secondary pathways were discovered. Comparisons with four previously prepared cDNA libraries revealed that this transcriptome dataset has both a high degree of consistency with previous EST data and an approximate 20 times increase in coverage. Thirteen unigenes related to theanine and flavonoid synthesis were validated. Their expression patterns in different organs of the tea plant were analyzed by RT-PCR and quantitative real time PCR (qRT-PCR).
Conclusions: An extensive transcriptome dataset has been obtained from the deep sequencing of tea plant. The coverage of the transcriptome is comprehensive enough to discover all known genes of several major metabolic pathways. This transcriptome dataset can serve as an important public information platform for gene expression, genomics, and functional genomic studies in C. sinensis.
Figures
References
-
- Yamamoto T, Juneja LR, Chu DC, Kim M, (Eds) Chemistry and Application of Green Tea. CRC Press, New York; 1998.
-
- Wang Y, Jiang CJ, Zhang HY. Observation on the Self-incompatibility of Pollen Tubes in Self-pollination of Tea Plant in Style in vivo. Tea Sci. 2008;28:429–435.
-
- Tanaka J, Taniguchi F. Estimation of the genome size of tea (Camellia sinensis), camellia (C. japonica), and their interspecific hybrids by flow cytometry. Journal of the Remote Sensing Society of Japan. 2006;101:1–7.
-
- Park JS, Kim JB, Hahn BS, Kim KH, Ha SH, Kim YH. EST analysis of genes involved in secondary metabolism in Camellia sinensis (tea), using suppression subtractive hybridization. Plant Sci. 2004;166:953–961. doi: 10.1016/j.plantsci.2003.12.010. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
